




Las parálisis periódicas son un trastorno poco frecuente que cursa con episodios de debilidad muscular aguda que puede confundirse con otras enfermedades como la epilepsia o la miastenia gravis, entre otras. Dentro de ellas se incluyen las parálisis hiper e hipopotasémicas, dividiéndose estas últimas a su vez en periódicas (familiar, tirotóxica o esporádica) y no periódicas. A este respecto, presentamos un caso de parálisis periódica hipopotasémica familiar en una mujer de 18 años que había sido diagnosticada en la infancia de epilepsia y que, además, había presentado un hipotiroidismo subclínico meses atrás por el que había recibido tratamiento sustitutivo. La anamnesis, siempre fundamental, y la objetivación de hipopotasemia permitieron el diagnóstico.
Periodic paralysis is a rare disorder that causes episodes of severe muscle weakness that can be confused with other diseases, including epilepsy or myasthenia gravis. Hyperkalemic and hypokalemic paralysis are included within these diseases, the latter being divided into periodic paralysis (familial, thyrotoxic or sporadic) and non-periodic paralysis. In this regard, we present a case of familial hypokalemic periodic paralysis in an eighteen year-old female who was diagnosed with epilepsy in childhood, as well as a subclinical hypothyroidism (for which she received replacement therapy) months ago. The diagnosis was made by the anamnesis and the confirmation of hypokalemia.
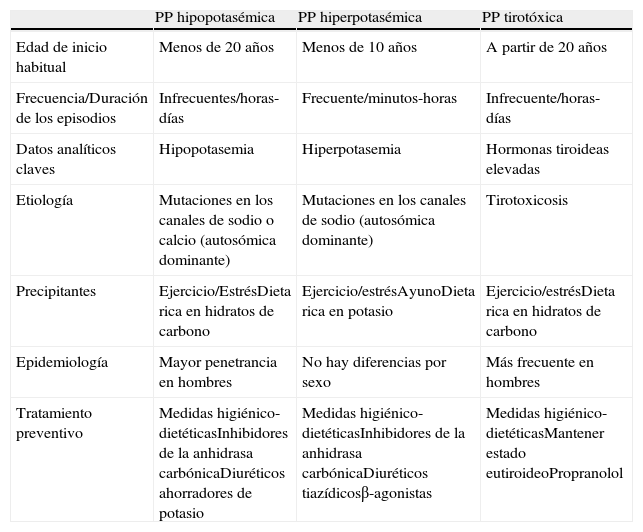
Las parálisis periódicas son un trastorno poco frecuente (prevalencia aproximada: 1 caso/100.000 habitantes)1, habitualmente transmitido de forma autosómica dominante (menor penetrancia en mujeres)2 y que cursa con episodios agudos de debilidad muscular. En cuanto a la clasificación de las crisis de parálisis, estas se pueden dividir, en función de los niveles de potasio durante los episodios, en hiperpotasémicas e hipopotasémicas, las cuales presentan algunas diferencias que se resumen en la tabla 1, aunque será la hipopotásemica la que nos ocupe en adelante.
Diagnóstico diferencial entre las parálisis periódicas hipopotasémica, hiperpotasémica y tirotóxica
| PP hipopotasémica | PP hiperpotasémica | PP tirotóxica | |
| Edad de inicio habitual | Menos de 20 años | Menos de 10 años | A partir de 20 años |
| Frecuencia/Duración de los episodios | Infrecuentes/horas-días | Frecuente/minutos-horas | Infrecuente/horas-días |
| Datos analíticos claves | Hipopotasemia | Hiperpotasemia | Hormonas tiroideas elevadas |
| Etiología | Mutaciones en los canales de sodio o calcio (autosómica dominante) | Mutaciones en los canales de sodio (autosómica dominante) | Tirotoxicosis |
| Precipitantes | Ejercicio/EstrésDieta rica en hidratos de carbono | Ejercicio/estrésAyunoDieta rica en potasio | Ejercicio/estrésDieta rica en hidratos de carbono |
| Epidemiología | Mayor penetrancia en hombres | No hay diferencias por sexo | Más frecuente en hombres |
| Tratamiento preventivo | Medidas higiénico-dietéticasInhibidores de la anhidrasa carbónicaDiuréticos ahorradores de potasio | Medidas higiénico-dietéticasInhibidores de la anhidrasa carbónicaDiuréticos tiazídicosβ-agonistas | Medidas higiénico-dietéticasMantener estado eutiroideoPropranolol |
Modificada de Gutman y Robin10.
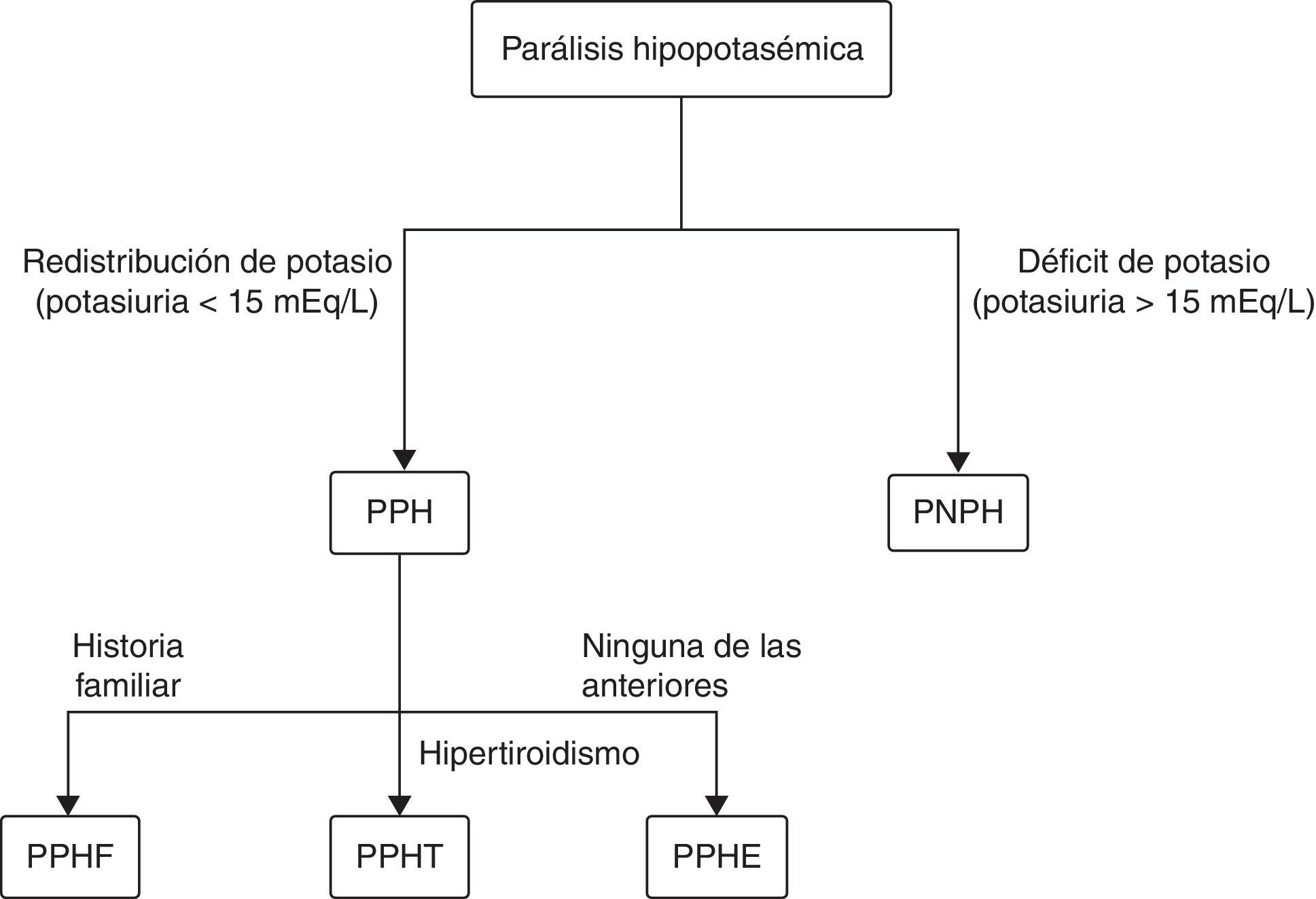
Las parálisis hipopotasémicas (PH) se dividen en periódicas (PPH) y en no periódicas (PNPH), siendo las primeras secundarias a la entrada de potasio al interior de la célula y las segundas consecuencia de un déficit de potasio. A su vez, las PPH se clasifican en familiar (PPHF), esporádica (PPHE) y tirotóxica (PPHT; tabla 1). Para diferenciar de forma básica los distintos tipos de parálisis hipopotasémicas puede ser de utilidad el algoritmo que se representa en la figura 1.
. PPH: Parálisis periódica hipopotasémica. PNPH: parálisis no periódica hipopotasémica; PPHF: parálisis periódica hipopotasémica familiar; PPHT: parálisis periódica hipopotasémica tirotóxica; PPHE: parálisis periódica hipopotasémica esporádica.")
Algoritmo diagnóstico básico de las parálisis hipopotasémicas. (Modificada de Frantchez et al.6).
PPH: Parálisis periódica hipopotasémica. PNPH: parálisis no periódica hipopotasémica; PPHF: parálisis periódica hipopotasémica familiar; PPHT: parálisis periódica hipopotasémica tirotóxica; PPHE: parálisis periódica hipopotasémica esporádica.
En diciembre de 2010 concurrió al servicio de urgencias del Hospital Virgen de Valme (Sevilla, España) una mujer de 18 años, gestante de 16 semanas, que acudía por presentar un episodio de tetraparesia que se presentó de forma brusca mientras descansaba la noche anterior y que no se acompañaba de ninguna otra sintomatología, respetándose además el nivel de conciencia, el lenguaje, el control de esfínteres y los movimientos oculocefálicos. Refería haber presentado episodios previos de parálisis similares (sin referir ningún desencadenante) pero que cedieron en pocas horas, por lo que el motivo de consultar en esta ocasión fue la persistencia de la clínica más tiempo del habitual. Negaba cualquier cambio en su medicación, en la alimentación o en la actividad física, aunque afirmaba que en los días previos estaba presentando vómitos copiosos.
Entre los antecedentes personales destacaba que estaba embarazada de 16 semanas, que estaba diagnosticada de hipotiroidismo subclínico, habiendo seguido tratamiento sustitutivo (aunque no lo hacía desde hacía unos 3 meses) y que en 1996 había sido diagnosticada (y tratada) de epilepsia tras ser estudiada por episodios transitorios de hipotonía generalizada. En cuanto a los antecedentes familiares, cabe mencionar la presencia, tanto en el padre de la paciente como en 7 familiares de este, de crisis de parálisis que cedían espontáneamente y que nunca fueron estudiadas.
En la exploración neurológica se observó una tetraparesia flácida, pudiendo la paciente mover, parcialmente, únicamente el miembro superior derecho. Además, mantenía un buen nivel de conciencia, los reflejos osteotendinosos estaban hipoactivos, estando los aquíleos abolidos y siendo el resto de la exploración neurológica normal. Por lo demás, la paciente presentaba un buen estado general, estando afebril y hemodinámicamente estable, sin que hubiese nada que destacar de la auscultación cardiopulmonar ni del resto de la exploración sistemática por aparatos.
Dada la situación, se solicitaron bioquímica, hormonas tiroideas, hemograma, coagulación, gasometría venosa y electrocardiograma; siendo todos los resultados normales salvo los niveles de potasio en sangre (K+ 2,4 mEq/l [3,5-4,5 mEq/l]). Ello motivó el inicio del tratamiento con potasio oral, estando la paciente al día siguiente completamente asintomática y siendo dada de alta con diagnóstico de episodio de parálisis periódica hipopotasémica familiar probablemente precipitado por hiperemesis aguda.
DiscusiónHay, a partir de este caso, 2 elementos que resulta interesante destacar. El primero de ellos es el diagnóstico de la enfermedad y, más concretamente, el diagnóstico diferencial de la misma. En cuanto al segundo elemento, merece la pena destacarlo por no haberse mencionado hasta ahora: el tratamiento.
En lo referente al diagnóstico, la debilidad muscular aguda, la exploración realizada y la objetivación de hipopotasemia orientaron de forma rápida hacia una PPH. Una vez en ese punto, es importante, además de distinguir el tipo de PPH ante el que nos encontramos, valorar las repercusiones que pueda estar originando el cuadro. Por ello, es necesario solicitar, además de una bioquímica y un hemograma, el nivel de hormonas tiroideas, una gasometría venosa y un electrocardiograma. En caso de dudas diagnósticas, se pueden realizar, personalizando en cada caso, otros estudios como el electromiograma, la biopsia muscular (baja especificidad) o las pruebas de provocación (que pueden ser arriesgadas) con glucosa y/o insulina, ejercicio o ACTH.
A pesar de todo, es importante recordar la necesidad y utilidad, como siempre, de una anamnesis cuidadosa que permita una mejor orientación diagnóstica. En este caso, la presencia de múltiples episodios similares tanto en la paciente como en sus familiares nos acercan casi con toda seguridad al diagnóstico de parálisis periódica hipopotasémica familiar, lo cual podría confirmarse mediante un estudio genético (en un 70% de los pacientes con PPH existe una mutación en el gen que codifica la subunidad alfa-1 de los canales de calcio del músculo esquelético, aunque también se han detectado otras mutaciones menos frecuentes como, por ejemplo, en los canales de sodio SCN4A)3–5.
Por otro lado, llama la atención en este caso que la paciente hubiese sido diagnosticada y tratada de epilepsia (trastorno que no padece) por episodios similares a este. Así pues, ante episodios de debilidad muscular aguda se debe incluir en el diagnóstico diferencial a la PPHF (la cual se podrá confirmar con una buena anamnesis y los estudios complementarios ya descritos), junto con otras enfermedades con las que podría confundirse: parálisis periódicas tirotóxica o hiperpotasémica, epilepsia, miastenia gravis, miopatías metabólicas, hipopotasemias secundarias, Guillain-Barré, mielopatía aguda o botulismo entre otras.
En lo que al tratamiento se refiere, este se basa principalmente en el tratamiento agudo del episodio y en el posterior tratamiento preventivo de nuevos episodios. En el tratamiento agudo las prioridades deben ser prevenir posibles arritmias (alargamiento del QT, aumento de onda U) o un posible fallo respiratorio, para lo cual se deberá realizar una monitorización cardiorrespiratoria, y revertir la parálisis mediante la administración de potasio para recuperar los valores normales6. Dicha administración podría realizarse administrando 30 mEq de ClK vía oral cada 30 min hasta la normalización7, aunque, debido al riesgo de hiperpotasemia de rebote, una vez que se produzca la salida de potasio al exterior de la célula, hay quien recomienda una administración más lenta (10 mEq de ClK vía oral cada 60 min)8,9. También puede utilizarse la vía intravenosa, indicada especialmente ante niveles de potasio en sangre menores de 2,5 mEq/l6 y/o si el paciente es incapaz de deglutir. Además, no debemos olvidar, en el tratamiento agudo, que no deben administrarse sueros glucosilados, puesto que provocarán un aumento de insulina que agravará la hipopotasemia.
Por último, en lo referente al tratamiento profiláctico, es importante destacar que las medidas no farmacológicas son habitualmente suficientes. En estas medidas se incluyen fundamentalmente una dieta rica en potasio y baja en hidratos de carbono y evitar el ejercicio excesivo. En el caso de que estas medidas no fueran suficientes, podrían utilizarse distintos fármacos10, de entre los que destacan fundamentalmente los suplementos de potasio, los diuréticos ahorradores de potasio (espironolactona 100mg/día, triamtereno 150mg/día) y los inhibidores de la anhidrasa carbónica (acetazolamida 250mg/12 h, que puede combinarse con espironolactona7). Además, se está aún estudiando la eficacia del verapamilo (240mg/día) y del topiramato (75-100mg/12 h), existiendo datos de que este último podría disminuir la severidad de los ataques, aunque no su frecuencia.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
