




La poliquistosis renal dominante del adulto, aunque es la enfermedad hereditaria más frecuente en la población española, presenta una prevalencia muy baja. Los síntomas iniciales son generalmente inespecíficos y difíciles de detectar por el médico. El método de elección para el diagnóstico de la enfermedad, cuando esta es asintomática, es la ecografía.
Presentamos un caso clínico poco frecuente, en el cual el diagnóstico de poliquistosis renal complicada se realiza de forma incidental durante el estudio de un cuadro que simulaba una colecistitis aguda. Queremos resaltar la importancia que tienen una anamnesis detallada y metodológica en el diagnóstico precoz de esta enfermedad.
Adult polycystic kidney disease, although it is the most common hereditary disease in the Spanish population, has very low prevalence. Initial symptoms are usually nonspecific and difficult to detect by the physician. The ultrasonography is the method of choice for diagnosing when the disease is asymptomatic.
We report a rare clinical case in which complicated polycystic kidney disease was diagnosed by chance during the study of a simulated acute cholecystitis. We emphasize the importance of making a detailed and methodological medical history for the early diagnosis of this disease.
La poliquistosis renal dominante del adulto es la enfermedad hereditaria más frecuente en la población española, con una frecuencia aproximada de 1/1.000 individuos1.
Los síntomas iniciales son inespecíficos y frecuentemente difíciles de detectar por parte del médico. La mayor parte de los pacientes mayores de 30 años presentan quistes renales. La evolución y gravedad de la enfermedad, incluso entre los individuos de una misma familia es muy variable. En la fase terminal los riñones afectados claudican, por lo que la diálisis y el trasplante renal son las únicas alternativas terapéuticas.
El diagnóstico precoz de esta enfermedad es fundamental para instaurar sin demora el tratamiento adecuado y, así, disminuir la progresión de la enfermedad. El método de elección para el diagnóstico de la poliquistosis renal, cuando esta es asintomática, es la ecografía2.
Caso clínicoVarón de 28 años de edad que acude a Urgencias por presentar un cuadro de dolor sordo y continuo en hipocondrio derecho, no irradiado, de características inflamatorias de 3 días de evolución. Entre sus antecedentes personales no presenta patologías crónicas de interés. Su madre falleció por un proceso crónico de origen renal de causa desconocida por el paciente. A la exploración, se encuentra afebril, con una tensión arterial de 140/85mmHg, presentando dolor leve a la palpación profunda en el hipocondrio derecho. Los signos de Murphy, Courvoisier y el resto de la exploración clínica son negativos. Con la sospecha inicial de cólico biliar, se realiza un hemograma con bioquímica urgente y se inicia tratamiento sintomático con antiinflamatorios no esteroideos y N-butilbromuro de hioscina.
A las pocas horas se produce un evidente empeoramiento de los síntomas, con aumento del dolor, hipertermia y el signo de Murphy positivo. Los análisis sanguíneos revelan una ligera leucocitosis sin desviación a la izquierda, junto con un leve aumento de la creatinina.
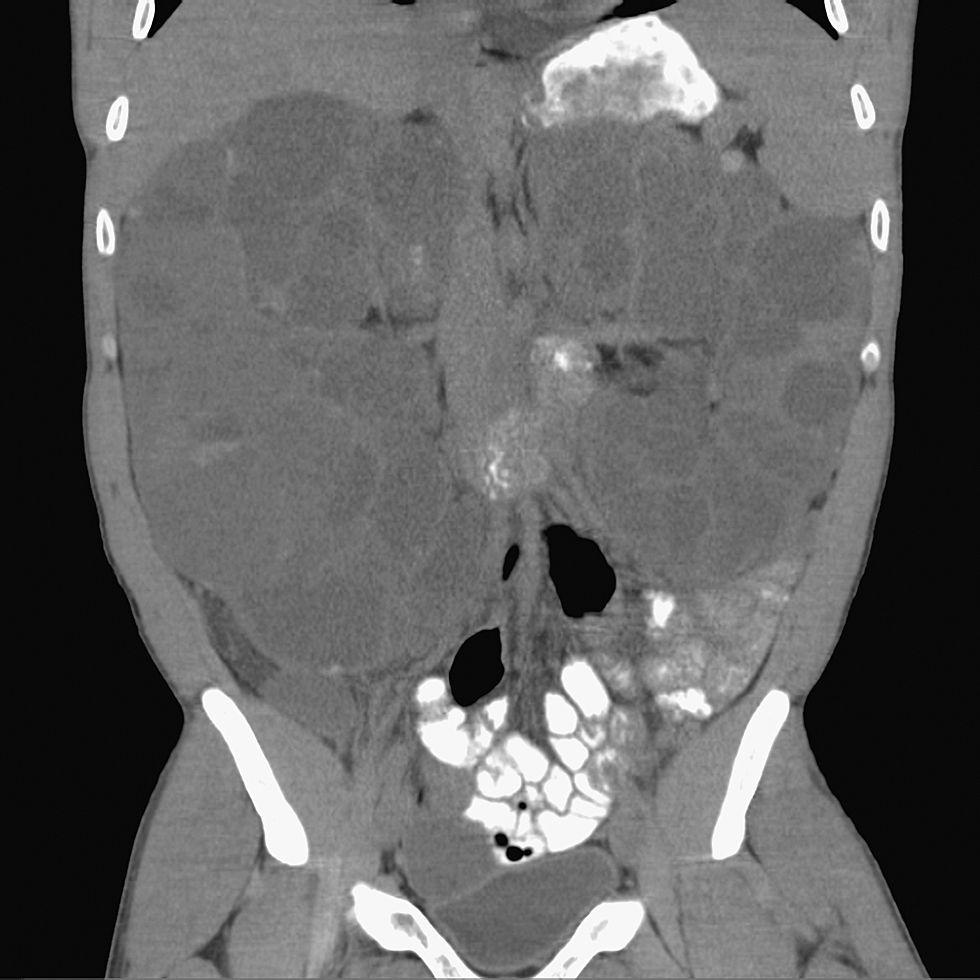
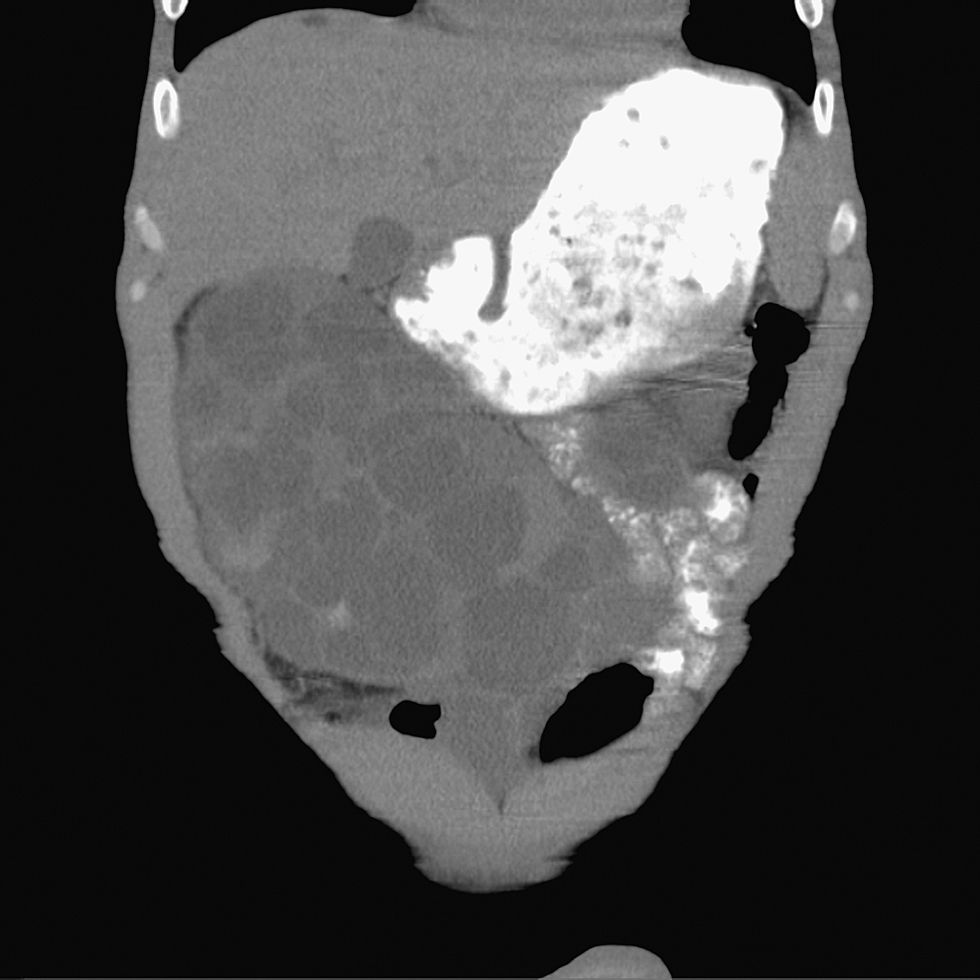
Ante la sospecha de colecistitis aguda, se solicita urgentemente una ecografía abdominal y se instaura de forma empírica tratamiento con amoxicilina‐clavulánico. La ecografía revela una vesícula biliar de características normales, desplazada medialmente por varios quistes dependientes del riñón derecho. El diagnóstico definitivo de poliquistosis renal complicada se realiza, finalmente, con la tomografía computarizada, donde se observan múltiples quistes renales bilaterales, algunos de los cuales se encuentran abscesificados (figs. 1 y 2).


El paciente es ingresado para recibir tratamiento intravenoso con antibióticos de amplio espectro, siendo dado de alta a los 7 días del ingreso, estando asintomático y con buen estado general.
DiscusiónLa variante autosómica dominante de la poliquistosis renal es una de las enfermedades hereditarias más frecuentes. Afecta a una de cada mil personas y se caracteriza por el desarrollo progresivo de quistes renales1. En ocasiones, se asocia a la poliquistosis hepática de adulto3.
Existen dos genes causantes, el gen PKD1, localizado en el cromosoma 16, y el gen PKD2, localizado en el cromosoma 44. El 85% de las poliquistosis renales autosómicas dominantes son causadas por el gen PKD1 y el resto, por mutaciones en el gen PKD2. La enfermedad asociada al gen PKD1 es más grave que la asociada al gen PKD2 con inicio en edades más tempranas5,6.
Como todas las enfermedades con herencia autosómica dominante, la presencia de una sola copia del gen mutado es suficiente para que la enfermedad se manifieste. Las principales características de este patrón de herencia son la transmisión vertical, en la que cada paciente tiene al padre o a la madre afectados por la enfermedad, el mismo riesgo para ambos sexos de padecer o transmitir la enfermedad y una probabilidad del 50% de que los descendientes estén afectados.
Generalmente, los síntomas iniciales de la poliquistosis renal son inespecíficos y difíciles de detectar, por lo que el papel del médico de Atención Primaria en las fases iniciales es clave para su diagnóstico7.
Debemos sospechar una poliquistosis renal en todo paciente con antecedentes familiares de patología crónica renal que presenten un cuadro de dolor lumbar y/o abdominal en presencia de masa, hematuria, hipertensión arterial y/o insuficiencia renal.
La mayoría de estos pacientes, cuando son diagnosticados, se encuentran en fases avanzadas y requieren un tratamiento especializado por parte del nefrólogo. El único objetivo del tratamiento es disminuir la progresión de la enfermedad mediante el control de la hipertensión arterial y de la insuficiencia renal.
Actualmente, existen estudios en animales de antagonistas de los receptores V2 de la vasopresina que parecen disminuir el crecimiento de los quistes.
Aproximadamente, la mitad de estos pacientes progresan rápidamente hacia la insuficiencia renal terminal necesitando, como última alternativa terapéutica, la diálisis y el trasplante renal8.
ConclusiónLos síntomas iniciales de la poliquistosis renal son inespecíficos y frecuentemente difíciles de detectar por parte del médico, por lo que una anamnesis detallada junto con una exploración exhaustiva y metodológica, así como el estudio mediante las técnicas precisas, son la base para su diagnóstico.
Se trata de una enfermedad poco frecuente y desconocida para muchos facultativos, por lo que es esencial conocer su existencia con la finalidad de realizar un diagnóstico precoz y de conocer las implicaciones que la enfermedad supone para el paciente.
