







Las prionpatías o encefalopatías por priones son un grupo de enfermedades poco frecuentes que comparten una fisiopatología similar con distintas características clínicas. La enfermedad de Creutzfeldt-Jakob (ECJ) esporádica es la más conocida. Se manifiesta con una demencia rápidamente progresiva, ataxia y sintomatología extrapiramidal. Aunque el diagnóstico de certeza es anatomopatológico se puede llegar al diagnóstico probable empleando los criterios establecidos por la Organización Mundial de la Salud. Se está discutiendo en la actualidad la posibilidad de integrar la resonancia magnética nuclear en dichos criterios para aumentar la sensibilidad del diagnóstico. Las técnicas moleculares de estudio de proteínas en el líquido cefalorraquídeo tienen un peso creciente y colaboran en el diagnóstico. El diagnóstico de las otras encefalopatías por priones no es tan avanzado como el de la ECJ. El tratamiento de todas estas enfermedades continúa siendo paliativo.
Prion diseases or prion encephalopathies are a group of rare disorders that share a similar pathophysiology with different clinical characteristics. Sporadic Creutzfeldt-Jakob disease (CJD) is best known. It presents as a rapidly progressive dementia, ataxia and extrapyramidal symptoms. Although accurate diagnosis is by histopathological examination, a diagnosis can be probably achieved using the criteria established by the World Health Organization. We are currently discussing the possibility of integrating the nuclear magnetic resonance into this criteria for increasing the sensitivity of diagnosis. Molecular techniques for studying proteins in cerebrospinal fluid have an increasingly significant role in aiding diagnosis. The diagnosis of other prion encephalopathies is not as advanced as that of CJD. The treatment of these diseases remains palliative.
Se denomina prión a la forma alterada de una proteína celular funcional (PrP en mamíferos) que ha podido perder su función normal pero que ha adquirido la propiedad de transformar la forma normal en patológica. Esta proteína tiene una conformación normal denominada PrPc, codificada por un gen (PRNP) que en humanos se encuentra en el cromosoma 20. En las patologías por priones aparece una isoforma alterada de la PrPc, denominada PrPsc que tiende a la formación de agregados de amiloide.
Los priones se reproducen en los mamíferos y resisten su inactivación al modificar los ácidos nucleicos, lo que les permite infectar a otras células. La secuencia primaria de aminoácidos de las dos isoformas es idéntica.
Las prionpatías o enfermedades por priones son un grupo de enfermedades neurodegenerativas de etiología idiopática, adquirida o genética (tabla 1). Son trastornos de la conformación de las proteínas, que se manifiestan como encefalopatía espongiforme en animales y como enfermedades neurodegenerativas en los humanos1. La acumulación de PrPsc produce afectación de la sustancia gris con muerte neuronal, gliosis y cambios espongiformes. El denominado amiloide priónico se deposita en forma de placas en el encéfalo, y se origina como consecuencia de una proteólisis incompleta de la PrPsc. La proteína priónica (PrP) está codificada por el gen de la proteína priónica (PRNP) del brazo corto del cromosoma 20 cuyo polimorfismo marcará el tipo de enfermedad por priones que padece el paciente2. Las enfermedades por priones tienen en común mutaciones en distintos codones del gen PRNP. La penetrancia de dichas mutaciones es variada en la población y las manifestaciones clínicas pueden ser muy heterogéneas3.
Clasificación de las prionpatías
| Clásica: |
|
| Adquiridas (PrPsc exógeno): |
|
| Genéticas (mutaciones del gen PRNP): |
|
PRNP: gen que codifica la proteína priónica; PrP: forma celular normal de la proteína priónica.
Según el Registro Nacional de Encefalopatías Espongiformes Transmisibles Humanas4, en España se han diagnosticado 1.163 casos de enfermedades por priones desde 1993 hasta octubre de 2009. La más frecuente (934 pacientes) ha sido la enfermedad de Creutzfeldt-Jacob (ECJ). Dentro de este grupo, casi todos los casos fueron por la variante esporádica (885 casos registrados) y solo 6 casos derivados de implantes de duramadre realizados antes de 1989 (ECJ iatrogénica). En nuestro país, el diagnóstico se realiza con mayor frecuencia en los 60 años en varones y 70 años en mujeres.
Enfermedad de Creutzfeldt-JakobLa enfermedad de Creutzfeldt-Jakob5 es una rara encefalopatía transmisible producida por priones. Los individuos afectados pueden presentar alteraciones del sueño, cambios de personalidad, ataxia, afasia, pérdida de la visión, debilidad, atrofia muscular, mioclonías y demencia progresiva hasta la muerte.
Se describen tres formas de presentación (tabla 1): ECJ esporádica o clásica (85–90%), con una incidencia mundial de 1/1.000.000; ECJ familiar (10–15%), con herencia autosómica dominante producida por una mutación de la línea germinal del gen PRNP, y heredándose de esta forma la proteína anormal; ECJ trasmitida o iatrogénica (1%), por inoculación accidental de priones (uso de hormona de crecimiento, transplantes corneales e injertos de duramadre contaminados).
La ECJ presenta una incidencia mayor en varones, con una supervivencia de unos 5 meses, siendo discretamente más elevada en mujeres. Se asocia a un peor pronóstico si existen signos típicos en el electroencefalograma (EEG), homocigosis en el codón 129 metionina/metionina (M/M) y con la presencia de la proteína 14.3.3 en el líquido cefalorraquídeo (LCR)6.
El diagnóstico de ECJ es habitualmente difícil de realizar. La edad media de aparición de la forma clásica es entre los 57 y los 62 años. El diagnóstico definitivo solo puede realizarse post mortem tras la necropsia. El diagnóstico de presunción se fundamenta en la clínica y en pruebas de laboratorio (tablas 2 y 3):
- A)
Clínica7. Un tercio de los pacientes presenta síntomas prodrómicos inespecíficos, como fatiga, trastornos del sueño, disminución de peso, cefaleas, malestar y dolor mal definido. Posteriormente se desarrolla una demencia rápidamente progresiva, ataxia, descoordinación, mioclonías (90%), disfunción extrapiramidal (rigidez, temblor, facies de máscara, movimientos coreoatetósicos), crisis epilépticas (generalmente crisis motoras generalizadas desencadenadas habitualmente por estímulos auditivos o táctiles) y signos vegetativos (cambios en el peso corporal, en la temperatura, sudoración). En general, los pacientes diagnosticados antes de los 50 años presentan más clínica extrapiramidal y psiquiátrica, mientras que los pacientes con un diagnóstico más tardío presentan demencia como signo principal.
- B)
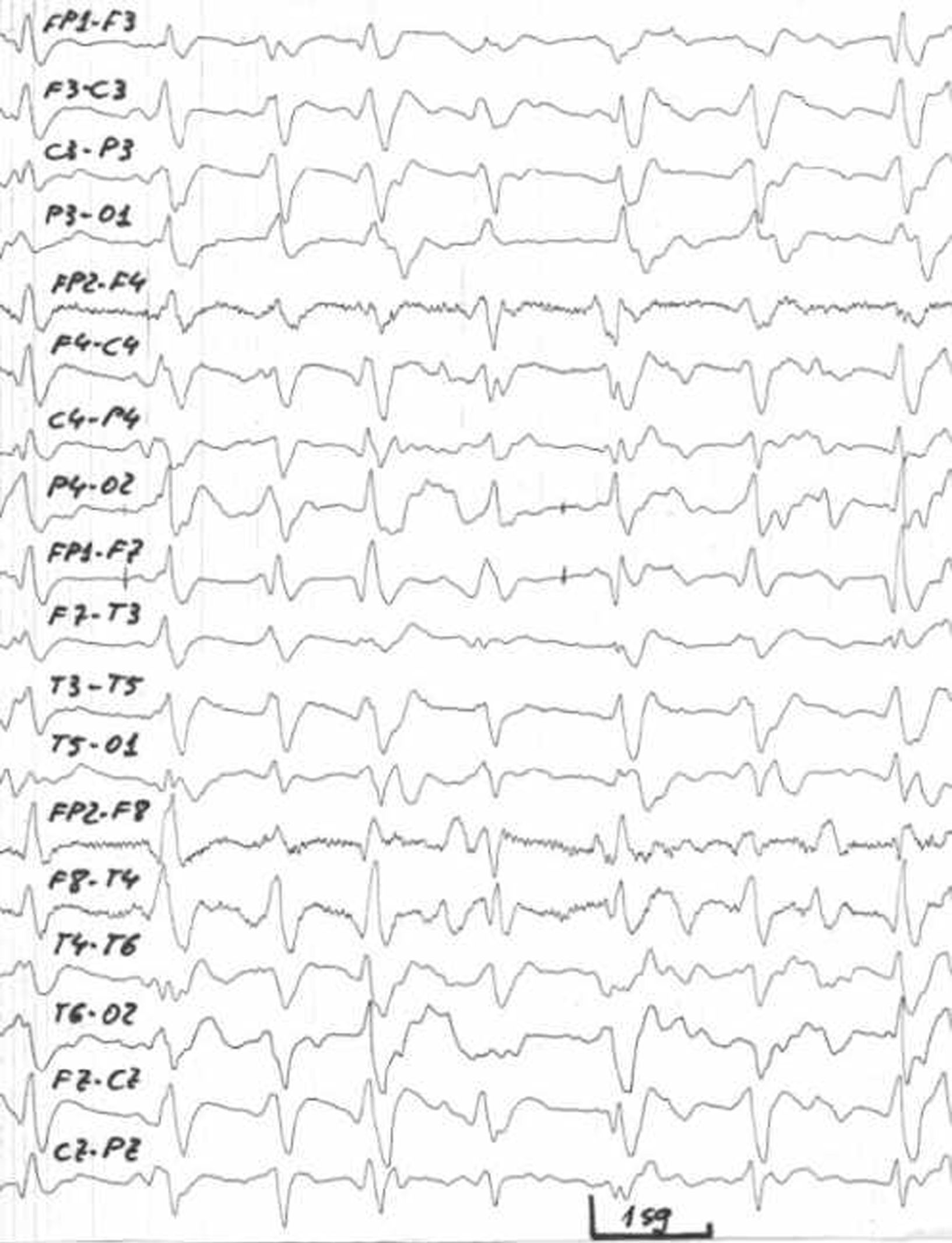
Electroencefalograma8. En el inicio de la enfermedad se observa una lentificación difusa de la actividad de fondo, aunque también es posible encontrar anomalías focales. Posteriormente aparecen descargas periódicas de ondas agudas, aunque de manera intermitente o esporádica, que pueden desencadenarse selectivamente por estímulos aferentes. El trazado típico (fig. 1) es una actividad delta sinusoidal con grafoelementos escarpados difásicos o trifásicos, de voltaje elevado (100–150μV) de aproximadamente 100–300 milisegundos de duración, en forma de brotes rítmicos intermitentes (0,7–1,5/segundo) de predominio frontal, conocidos como frontal intermittent rhythmic delta activity (FIRDA). Según progresa la disminución del nivel de conciencia los complejos periódicos de ondas son sustituidos por el patrón alternante cíclico (PAC) con una fase de descarga sostenida de complejos periódicos y otra fase con actividad theta-delta rítmica acompañada de decremento en la actividad muscular y de la frecuencia cardiorrespiratoria. La sensibilidad de esta técnica no es muy alta (42%) sin embargo tiene una alta especificidad (92%).
- C)
Líquido cefaloraquídeo9. Generalmente es normal, siendo frecuente una leve elevación de las proteínas (<100mg/dl). La proteína 14.3.3 forma parte de una familia de proteínas con posibles funciones reguladoras celulares que se liberan en el LCR en respuesta a una destrucción extensa del tejido cerebral. Según el Registro Nacional de Encefalopatías Espongiformes Transmisibles Humanas4, en España ha sido estudiada en el 90% de los pacientes aunque está presente en menos del 10% de los casos diagnosticados. El test de detección por Western Blot ofrece una sensibilidad superior al 90% y especificidad del 87%. Esta proteína también puede detectarse en síndromes paraneoplásicos, ictus, meningoencefalitis, encefalopatías hipóxico-metabólicas, metástasis y otras demencias10. A pesar de su presencia en el LCR no permite distinguir la ECJ de otras demencias rápidamente progresivas11. Otras proteínas presentes que también han sido estudiadas son tau, S-100b, enolasa neuronal, tau y abeta fosforiladas12.
- D)
Pruebas de imagen. En la resonancia magnética nuclear (RMN) craneal13 se pueden ver áreas de hiperdensidad en núcleo caudado, globus palidus y tálamo, que posiblemente reflejan la extensión de la espongiosis y la degeneración. El signo más específico es el aumento de señal en el núcleo caudado y el putamen de forma simultánea. Aún así debe considerarse el diagnóstico si la hiperintensidad afecta al menos a dos territorios cerebrales (temporal, parietal u occipital). Algunos autores14 sugieren que deben incluirse los parámetros de la RMN y la proteína 14.3.3 del LCR en los criterios diagnósticos propuestos por la OMS15 (tablas 2 y 3). Al incluir estos criterios, se alcanza una sensibilidad diagnóstica del 98,8, frente al 91,8% que ofrecen los criterios de la OMS15. En la tomografía por emisión de positrones (PET) y en la tomografía computarizada por emisión de positrones individuales (SPECT) pueden verse áreas inespecíficas de anormal metabolismo y perfusión.
- E)
Análisis genético de la proteína criónica. Destaca un polimorfismo para M/M en el codón 129 del gen PRNP en el 67% de los pacientes con ECJ esporádica y en el 69% de los pacientes con ECJ familiar. El siguiente polimorfismo más frecuente es el formado por metionina/valina (M/V)2. En España16, el polimorfismo M/M se estima en el 52%, y el M/V en un 35%.
- F)
Anatomía patológica17. Se evidencia una degeneración espongiforme en el sistema nervioso central (aparición de vacuolas de 1–5μm entre los cuerpos neuronales de corteza cerebral, putamen, núcleo caudado, tálamo y capa molecular de cerebelo), astrogliosis en la sustancia gris, depósito de placas de amiloide (10–15%), y típica ausencia de respuesta inflamatoria. La presencia de fibrillas asociadas a scrapie (SAF) junto con la detección de la resistencia a proteasa de la PrPsc, constituyen una buena evidencia diagnóstica.
.")
Criterios diagnósticos de la enfermedad Creutzfeldt-Jakob según la OMS (2003)15
| ECJ definitiva |
|
| ECJ probable |
|
| ECJ posible |
|
ECJ: enfermedad de Creutzfeldt-Jacob; EEG: electroencefalograma; PrP: forma celular normal de la proteína priónica.
Criterios diagnósticos de la ECJ propuestos por Zerr et al (2009)14
| ECJ confirmada |
|
| ECJ probable | Al menos 2 criterios clínicos y 1 test diagnóstico |
| ECJ posible | Al menos 2 criterios clínicos y duración menor de 2 años |
| I: Criterios clínicos: | |
| |
| II: Test diagnósticos: | |
| |
ECJ: enfermedad de Creutzfeldt-Jacob; EEG: electroencefalograma; LCR: líquido cefaloraquídeo; PrP: proteína priónica; RMN: resonancia magnética nuclear.
Aparece en pacientes jóvenes (edad media de 28 años) que presentan homocigosis para el codón 129 de la PRNP18. Se transmite por la ingesta de carne bovina procedente de ganado enfermo con encefalopatía espongiforme bovina. Su evolución es letal en un período de 6 a 24 meses. Se inicia con clínica psiquiátrica (ansiedad, depresión e incluso psicosis), seguida por parestesias difusas (en cara, miembros superiores e inferiores y en algunos casos todo el hemicuerpo). La progresión de la enfermedad es rápida, con clínica neurológica en forma de ataxia seguida por movimientos anómalos, demencia, inmovilismo y mutismo. La RMN se caracteriza por presentar dos signos típicos: hiperseñal en pulvinar y en tálamo dorsomedial. El EEG es anormal al inicio con un patrón de onda lenta sin parecerse a la ECJ, aunque al final presentan características comunes. En anatomía patológica se observan «placas floridas» (núcleo central de amiloide de PrP rodeado de vacuolas con distribución en pétalos de flor) con cambios espongiformes en cerebro, cerebelo, ganglios basales y tálamo.
KuruEs una enfermedad por priones descrita entre los nativos de las tierras altas de Nueva Guinea producida por priones similar a la vECJ que se transmite por la ingesta de encéfalo y carne humana19. Como la práctica del canibalismo ritual ya no es habitual, los últimos casos diagnosticados fueron en los años 90. La enfermedad afecta principalmente a mujeres adultas y niños de ambos sexos. Se inicia con trastornos ansiosodepresivos que se acompañan de ataxia, aparición subaguda de disdiacocinesia, mioclonías, coreoatetosis y fasciculaciones, seguido por la debilidad motora e incontinencia. En las fases finales de la enfermedad aparece disartria, demencia y encamamiento. La vida media desde el diagnóstico oscila entre 9 y 24 meses. El EEG es anómalo pero no es similar al de otras enfermedades priónicas. Se ha observado una homocigosis en el mismo codón de la PrP que la vECJ. En la biopsia cerebral, se observa pérdida no inflamatoria neuronal, hipertrofia de los astrocitos, vacuolización espongiforme y placas de Kuru que contienen material amiloide de proteína priónica.
Insomnio familiar letalEs un trastorno autosómico dominante causado por una mutación en la proteína del prión20. Presenta un amplio rango de aparición entre los 23 y los 73 años. El cuadro se inicia y caracteriza por el insomnio, con pérdida del ritmo circadiano y alteraciones cognitivas (disminución de la atención, la concentración y la memoria). La polisomnografía muestra una alteración marcada del ritmo del sueño. Se acompaña de una disfunción autonómica caracterizada por hipertensión arterial, taquicardia, hipertermia e hiperhidrosis. Presenta una disminución en la secreción de hormona adrenocorticotropa (ACTH), un aumento del cortisol basal y una pérdida de la secreción circadiana de la hormona del crecimiento, melatonina y prolactina. La enfermedad progresa con mioclonías, ataxia y espasticidad. El PET muestra un característico descenso de la captación de glucosa en el tálamo. En la anatomía patológica se observa gliosis, degeneración espongiforme y disminución neuronal en tálamo.
Síndrome de Gerstmann-Straüssler-ScheinkerPresenta una incidencia de 1–10/1019 habitantes-año20. Tiene un patrón de herencia autosómico dominante en un 40% de los casos. La aparición clínica ocurre de la tercera a la sexta década de la vida y progresa hasta la muerte en un período aproximado de 5 años. Se han descrito algunos grupos emparentados con características clínicas y patológicas variables. El cuadro se inicia con debilidad proximal en miembros inferiores junto con hiporreflexia y disestesia. Se caracteriza por torpeza motora, disdiacocinesia, paraparesia espástica, signos extrapiramidales, y ataxia que evoluciona hacia una degeneración del tracto corticoespinal. La demencia no está siempre presente aunque es frecuente la agresividad, la labilidad emocional y la apatía. El EEG destaca con complejos onda punta similar al de la vECJ. La RMN muestra una discreta hiposeñal en T2 (tiempo de relajación transversal). Lo más sensible y específico para el diagnóstico es la mutación del gen PRNP. La anatomía patológica muestra placas similares a las de Kuru con espículas radiadas y cambios microgliales.
Nueva prionpatía sensible a proteinasaSe han descrito21 cinco casos hasta la actualidad, la edad media de aparición son los 62 años con una supervivencia de 20 meses. Se acompaña de clínica psiquiátrica, ataxia, parkinsonismo y demencia. Se observa un EEG enlentecido y una degeneración espongiforme de la corteza, ganglios de la base y tálamo.
Manejo terapéutico de las prionpatíasLa duración media de la enfermedad es de cinco meses y al año del inicio fallecen el 80% de los pacientes23.
Es importante diferenciar las enfermedades por priones genéticas de otras patologías neurodegenerativas, particularmente la enfermedad de Alzheimer. La OMS recomienda hacer un estudio genético a todos los pacientes con una historia familiar de enfermedad por priones. Las obvias consideraciones éticas de tal determinación hacen obligatorio obtener por parte del clínico el consentimiento informado del paciente o tutor del mismo.
Se han desarrollado medidas para evitar la transmisión de la enfermedad. La transmisibilidad e infectividad de tejidos y/o fluidos ha sido establecida por la OMS24 y los divide en cuatro categorías por orden decreciente. Los tejidos prohibidos como material específico de riesgo (MER) son el cerebro, la médula espinal, los ojos, las amígdalas y los intestinos. Junto a esto, el otro riesgo a tener en cuenta es la posibilidad de transmisión de la enfermedad de persona a persona a través de las prácticas médicas y quirúrgicas que se realizan. Por eso, además de los problemas de seguridad alimentaria, contaminación del medio ambiente, cosméticos, medicamentos y vacunas, se han tomado medidas a fin de minimizar el riesgo de transmisión por productos sanguíneos y por instrumental médico y quirúrgico23,25.
No hay tratamiento curativo en la actualidad. Se han empleado distintas líneas terapéuticas (aciclovir, amantadita, vidarabina, quinacrina, pentosano, anfotericina, interferón, clomipramina, levetiracepam, topiramato, fenitoína, flupirtine) pero no existe evidencia de que alguno de ellos mejore la supervivencia o controle los síntomas22. Hoy en día el tratamiento es únicamente paliativo.
ConclusionesLas encefalopatías por priones son enfermedades poco frecuentes en la práctica clínica de atención primaria pero deben considerarse en el diagnóstico diferencial de las demencias. Todas las prionpatías presentan como síntomas principales el deterioro cognitivo y la ataxia, y en ocasiones pueden presentar también sintomatología psiquiátrica. La clínica de la ECJ se caracteriza por demencia, ataxia, mioclonías y síntomas extrapiramidales. El diagnóstico de certeza solo se precisa con la necropsia. El diagnóstico probable requiere la realización de RMN, EEG y el análisis de proteínas en el LCR. Al no haber tratamiento curativo, el médico de familia desempeña un importante papel en los cuidados paliativos y manejo integral del paciente.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
