







La distrofia muscular de Duchenne, DMD* (CIE-9-C: 359.1; CIE-10-ES: G71.01, ORPHA: 98896) es una miopatía de tipo distrófico, autosómica recesiva ligada al cromosoma X, de baja incidencia 1/3300, con penetrancia completa y afectación multiorgánica (neuromuscular, respiratorio, digestivo y metabólico). Tiene una gran variabilidad clínica. Los síntomas se inician en la edad pediátrica (limitación de la movilidad y complicaciones respiratorias precoces). Las complicaciones respiratorias reducen la esperanza de vida de los afectados. No existe tratamiento que modifique su evolución, si bien los esteroides y las nuevas terapias génicas están aumentando la vida media de esta enfermedad. La función del médico de Atención Primaria es decisiva en el seguimiento y en el control de las complicaciones de la DMD, coordinando las diferentes especialidades implicadas en el mismo.
Duchenne muscular dystrophy, DMD*(ICD-9-C: 359.1; ICD-10-ES: G71.01, ORPHA: 98896) is a dystrophic type, autosomal recessive myopathy linked to the X chromosome, low incidence 1/3300, with full penetrance and multi-organ involvement (neuro-muscular, respiratory, digestive and metabolic). It has great clinical variability. Symptoms begin in pediatric age (mobility limitation and early respiratory complications). Respiratory complications reduce the life expectancy of those affected. There is no treatment that modifies its evolution, although corticosteroids and new gene therapies are increasing the half-life of this disease. The role of the Primary Care Physician (PCP) is decisive in the monitoring and control of the complications of DMD, either coordinating the different specialties involved in it.
La distrofia muscular de Duchenne (DMD) es una miopatía caracterizada por retraso del desarrollo motor debido a debilidad muscular proximal. La mayoría de los pacientes son diagnosticados en torno a los 5 años de edad. La esperanza de vida media es de 19 años y las complicaciones respiratorias recurrentes la principal complicación. Es considerada una enfermedad rara, con transmisión recesiva ligada al cromosoma X (son portadoras las mujeres y enfermos los hombres)1,2. El diagnóstico de sospecha se establece por los síntomas, antecedentes familiares y pruebas de laboratorio (elevación de transaminasas y creatinfosfocinasa [CK]). La confirmación diagnóstica requiere de un estudio molecular con PCR. El consejo genético debe de realizarse en los padres, siendo recomendable el diagnóstico prenatal. Actualmente, no existe tratamiento específico para esta entidad que permita detener o enlentecer su progresión. El tratamiento se basa en medidas de soporte de las complicaciones que aparecen de forma progresiva3. El paciente con DMD ha de ser evaluado y seguido por un equipo multidisciplinar y multiprofesional2. Aunque existe un soporte asociativo nacional importante (Duchenne Parent Project España), este es desigual en los territorios. La evolución de la enfermedad suele ser rápida y degenerativa; sin embargo, los tratamientos que están apareciendo y la presencia de mejores condiciones de fisioterapia y rehabilitación están aumentado la supervivencia más allá de la tercera década2,4.
Los Centros de Atención Primaria (CAP) suelen ser el primer punto de contacto con el sistema de atención de la salud para los pacientes con DMD5. Aunque la DMD se manifiesta en la infancia, dadas las particularidades de la asistencia sanitaria en España, la Atención Primaria (AP) ocupa un puesto importante ante el déficit de pediatras de AP. Los pacientes suelen acudir a los puntos de urgencia de AP y hospitalaria, en los que los profesionales sanitarios tienen poca experiencia en el tratamiento de esta afección5-7. Con este trabajo, se pretende brindar a los profesionales de AP y de Atención Urgente un acercamiento a los problemas comunes que afectan a los pacientes con DMD. El objetivo es que los profesionales de AP puedan conocer los recursos que tienen a su disposición para tratar las complicaciones médicas multisistémicas de la DMD: inmunización oportuna, asesoramiento de seguridad anticipada, detección del comportamiento y evaluaciones rutinarias de nutrición y desarrollo, así como el manejo cardiorrespiratorio, más incidente en este tipo de población. De esta manera, los profesionales de AP deben convertirse en un proveedor valioso y eficaz para los pacientes con DMD. Pueden ofrecer acceso y coordinación efectiva entre los cuidadores y los pacientes, y llegar a ser en un asesor de confianza para el paciente y su familia sobre decisiones médicas importantes, cuestiones en los ámbitos psicosocial, conductual y educativo5,7. La AP, como en otras enfermedades neurodegenerativas y crónicas en general, ha de convertirse en el referente aglutinador que sirva de conexión real entre niveles asistenciales.
Para dar a conocer un manejo actual e integrado en el ámbito de la AP se ha elaborado esta puesta al día DMD. Con la información de fondo que se analiza en este trabajo, tanto los profesionales de AP y urgencias pueden atender hábilmente a los pacientes con DMD en sus respectivos entornos, optimizando los resultados de los pacientes.
EpidemiologíaLa DMD es una enfermedad poco frecuente pero muy grave, con una afectación en España de 4,78/100.000 habitantes. Anualmente se diagnostican en el mundo unos 200.000 casos, siendo la incidencia de la DMD 1/3.300 nacimientos de varones4. Sin embargo, dado que los estudios de prevalencia e incidencia realizados para obtener estos datos en la DMD se basan en los test genéticos y las biopsias musculares realizados a pacientes con DMD, existen resultados contradictorios6. Debido a que existen pocos estudios en este sentido, y los resultados obtenidos son deficientes, estos hacen que sea difícil evaluar los posibles cambios en la forma en que se ha definido la DMD a lo largo del tiempo8.
Respecto a la mortalidad de la DMD, existe una tendencia mayor a la supervivencia, pero existen pocos estudios que hayan aportado nuevas evidencias sobre este hecho8. La mortalidad se ha visto reducida por la prescripción generalizada de corticoides, un mejor acceso a la ventilación mecánica no invasiva y la implementación de pautas de atención más completas y específicas9-11. También está demostrado que el método de diagnóstico está relacionado con la supervivencia. La realización de pruebas moleculares está asociada a una mortalidad más alta que cuando el diagnóstico es clínico en exclusiva11, ya que la incertidumbre en el diagnóstico afecta a la capacidad de estimar la prevalencia.
PatogeniaLa DMD es una enfermedad recesiva ligada al cromosoma X. Esta distrofia es una enfermedad de base genética como resultado de mutaciones (deleciones, fundamentalmente) en el gen de la distrofina (DMD: locus Xp21.2, el gen humano de mayor tamaño y que sufra una alta tasa de mutaciones endógenas), una proteína que está presente en el músculo esquelético, el músculo liso, el músculo cardíaco y el cerebro. Estas mutaciones conducen a la ausencia o defecto de dicha proteína1,4. La distrofina estabiliza la membrana celular de la fibra muscular esquelética en la contracción. Su ausencia determina que durante las contracciones musculares la transmisión de fuerzas desde el citosol a la matriz extracelular se vea alterada, conduciendo a la rotura de la membrana plasmática, produciéndose una entrada masiva de calcio extracelular y generando necrosis muscular (elevación de CK). Dada la respuesta miogénica, se produce un fenómeno de regeneración-degeneración muscular que favorece la fibrinogénesis. Por tanto, la biopsia muscular de la DMD refleja la proliferación de tejido conectivo, la regeneración de miofibras, la degeneración y la necrosis de fibras musculares y el infiltrado inflamatorio, sustituyéndose el tejido muscular por tejido adiposo5. Es precisamente esa base histopatológica la que va a condicionar las diferentes manifestaciones clínicas, especialmente en el ámbito cardiorrespiratorio.
Manifestaciones clínicasLa DMD está presente desde el nacimiento, manifestándose los primeros datos de debilidad muscular en torno al segundo y tercer año de vida7,12. Habitualmente, la marcha comienza más tardíamente que en un niño sano, con dificultad posterior para la carrera y el salto. Inicialmente, la falta de fuerza es más evidente en los músculos proximales frente a los distales, antes en miembros inferiores que en superiores4,5. De forma progresiva, el enfermo tiene dificultad para subir escalones y para levantarse del suelo, usando sus manos como apoyo para ello (maniobra de Gowers). En torno a los 6 años de edad se suele producir un acortamiento del tendón de Aquiles, con una marcha de puntillas, con seudohipertrofia de la musculatura gemelar y una postura lordótica. En la evolución se van sumando limitaciones funcionales y caídas, siendo necesario el uso de muletas y, finalmente, de sillas de ruedas6,7.
La debilidad de la musculatura, junto con la deformidad torácica por escoliosis, condicionan la dificultad respiratoria, predisponiendo a infecciones pulmonares graves, siendo la neumonía aspirativa una de las principales causas de muerte junto con dilatación gástrica aguda6.
Por último, es muy frecuente la presencia de miocardiopatía dilatada primaria y eventos arrítmicos, aunque la sintomatología es escasa y en etapas avanzadas de la enfermedad5.
Las manifestaciones clínicas más relevantes se resumen en la tabla 1.
Resumen de las manifestaciones clínicas más relevantes según evolución de la enfermedad
| Manifestaciones iniciales<5 años de vida | Manifestaciones intermedias> 5-7 años de vida | Manifestaciones graves> 7 <10 años vida | Manifestaciones muy graves> 10 años de vida |
|---|---|---|---|
| Maniobra de Gowers positivaDebilidad de miembros inferioresBalanceo de caderas en la marchaMarcha de puntillasAlteraciones de conducta y la comprensiónEscoliosis radiológica | Imposibilidad para el saltar y subir escalerasDificultad de la marchaInicio de escoliosis sintomáticaInicio de desnutriciónPrimeras manifestaciones iatrogénicas (esteroides) | Imposibilidad para la marchaImposibilidad para la bipedestaciónSíntomas moderados-graves de escoliosisInicio de manifestaciones respiratorias | Debilidad de miembros superioresNo sedestaciónNecesidad de ventilación mecánica no invasivaManifestaciones cardíacas, digestivas y respiratorias |
El diagnóstico de la enfermedad debe ser lo más precoz posible, con el objetivo de un inicio precoz de las intervenciones sobre el paciente. Muchos pacientes con enfermedades neuromusculares se identifican por primera vez durante sus visitas de rutina en pediatría11,12. Sin embargo, la evaluación del desarrollo no termina con la identificación de un trastorno neuromuscular, porque un paciente puede tener un problema de desarrollo concurrente, ya sea relacionado con la enfermedad neuromuscular subyacente o claramente no relacionado con la enfermedad. Por ejemplo, los niños con DMD tienen una mayor tasa de discapacidad intelectual, discapacidad de aprendizaje, trastorno del lenguaje, autismo y trastorno por déficit de atención/hiperactividad que los niños no afectados4,12.
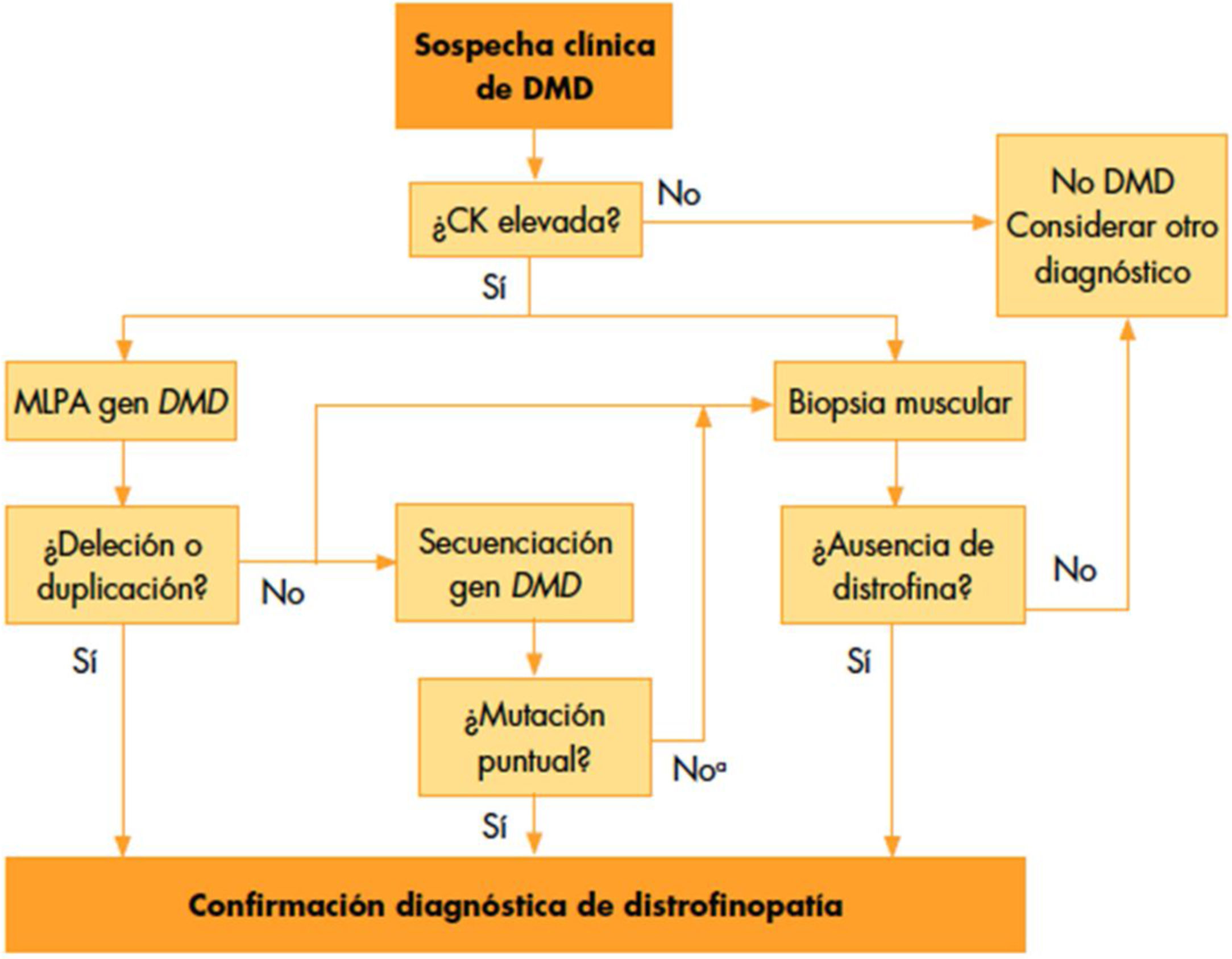
El enfoque inicial debe ser clínico, pudiéndose apoyar en algún hallazgo de laboratorio. Es típico de la DMD la elevación de la CK y las transaminasas. Las concentraciones séricas pueden estar elevadas en niños antes de la aparición de cualquier síntoma6,12. La CK puede alcanzar una elevación entre 10 y 100 veces su valor normal, con disminución progresiva según avanza la enfermedad (debido al reemplazo muscular por grasa y fibrosis en etapas finales). Una CK normal en fases iniciales debe hacernos pensar en otro diagnóstico, ya que la DMD sería muy poco probable5,6 (fig. 1).

Proceso diagnóstico en la DMD tomado de Camacho13.
Cuando la sospecha DMD sea alta, se recomienda hacer estudio genético. Si los resultados son positivos y la clínica es compatible, se puede establecer el diagnóstico de DMD. Dada la alta frecuencia de deleciones y duplicaciones, es razonable realizar primero pruebas genéticas de deleción/duplicación, como Multiplex Ligation-dependent Probe Amplification (MLPA). Si los resultados son negativos, debe secuenciarse el gen para buscar mutaciones puntuales o pequeñas deleciones/duplicaciones; caracterizando el error para evaluar su influencia en el estado del marco del fenotipo de la distrofinopatía. Si el estudio genético no detecta ninguna mutación, y la sospecha clínica es alta, se debe realizar una biopsia muscular para confirmar o descartar el diagnóstico, planteando diagnóstico diferencial con otras enfermedades5,6.
Aunque el estudio histopatológico actualmente tiene un segundo plano, la anatomía patológica revela en microscopia óptica un patrón distrófico con desestructuración de la arquitectura fascicular del músculo, necrosis y regeneración de fibras musculares, e incremento del tejido conectivo, con la sustitución del tejido muscular por tejido adiposo por el continuo ciclo de destrucción-regeneración5. Mediante inmunohistoquímica o inmunofluorescencia se comprueba la deficiencia de distrofina6.
Si se opta en primera instancia por realizar una biopsia del músculo (generalmente ha de biopsiarse un músculo no muy afectado) y se diagnostica la enfermedad comprobando el déficit de distrofina, a continuación, es obligatorio realizar las pruebas genéticas para identificar el tipo de mutación5,11.
La electromiografía no es una técnica diagnóstica, pero muestra cambios miopáticos, que generalmente consisten en pequeños potenciales polifásicos y se incluye siempre en el diagnóstico12.
Las pruebas de imagen radiológica no aportan hallazgos relevantes para el diagnóstico, quedando reservadas en el manejo de las complicaciones cardiorrespiratorias2,6,12.
El proceso diagnóstico se resume en el siguiente diagrama tomado de Camacho13.
Tratamiento de la distrofia muscular de DuchenneActualmente no se dispone de ninguna terapia curativa, por lo que es imprescindible el manejo multidisciplinar de estos pacientes y comenzar intervenciones precoces para retrasar la evolución de la enfermedad (tabla 2).
Tratamientos farmacológicos en DMD
| Tratamiento de base |
| Esteroides: valorar deflazacort a dosis de 0,9 mg/kg/día, aunque hay esquemas de prednisona y prednisolona válidos |
| Riesgo de desarrollo enfermedad cardíaca o enfermedad establecida: insuficiencia cardíaca o disfunción ventricular |
| Inhibidor de la enzima conversora de angiotensina |
| Antagonistas de los receptores de la angiotensina II |
| Betabloqueantes |
| Enfermedad de reflujo gastroesofágico |
| Inhibidor de la bomba de protones |
| Pacientes con mutación sin sentido en el gen de la distrofina |
| Valorar uso precoz de atalureno |
| Pacientes con mutación confirmada del exón 51 |
| Valorar uso de eteplirsen |
| Resto de pacientes sin mutación del exón 51 ni mutación del gen de la distrofina |
| Coordinación con el resto de especialidades para el manejo multidisciplinar y valorar por comité de expertos la inclusión en ensayos clínicos, si procede, según el perfil del paciente |
Es el único fármaco que ha demostrado beneficio. Los 2corticoides que se utilizan principalmente son la prednisona/prednisolona y deflazacort. No hay una fecha de comienzo ni una dosis estándar, aunque los 3 regímenes de uso más común son 0,75mg/kg/día de prednisona, 0,9mg/kg/día de deflazacort y 0,75mg/kg/día de prednisona en días alternos o por ciclos de 10 días (10 días de tratamiento con los próximos 10 días de descanso)11. Se ha constatado mejoría muscular y funcional, en la calidad de vida y beneficio tanto cardíaco como respiratorio5,11. En un metaanálisis reciente McDonald et al. comparan el estudio de tadalafilo DMD y el ACT DMD buscando la diferencia en la progresión de enfermedad entre los pacientes que toman deflazacort y prednisona/prednisolona. Aunque los estudios son difíciles de comparar y no tienen como objetivo primario valorar la diferencia entre ambas pautas de esteroides, se encuentra alguna pequeña diferencia estadísticamente significativa a favor de la pauta de deflazacort, con menor deterioro funcional a las 48 semanas de tratamiento14.
Hay que conocer este grupo farmacológico para prevenir sus efectos secundarios, como aumento de peso, síntomas gastrointestinales, cataratas u osteoporosis15.
Se debe individualizar su inicio según el paciente y su situación motriz, así como las dosis posteriores según la evolución de la enfermedad12,13. No obstante, el tratamiento debería comenzarse en las fases tempranas de la enfermedad, incluso antes de la aparición de los síntomas3.
Tratamientos no farmacológicosManejo musculoesquelético: el objetivo es la promoción de la movilidad amplia y simétrica participando para ello especialistas en enfermedad neuromuscular, rehabilitadores, fisioterapeutas y cirujanos ortopédicos. Para evaluar la efectividad del programa se pueden usar distintos test o escalas como el test de la marcha durante 6 min o la escala del Medical Research Council (tabla 3). El deporte es recomendable, evitando los de alta intensidad o traumáticos, y siendo muy beneficioso la natación. El uso de ortesis externas y cirugías ayudan a la corrección de deformidades y al mantenimiento de la movilidad articular y autonomía. En la fase no ambulatoria resulta vital la figura del terapeuta ocupacional2,5 (tabla 4).
Escala del Medical Research Council
| Grado 0. No se visualiza ni se palpa ninguna contracción |
| Grado 1. Leve contracción visible o palpable, aunque no se observa movimiento de la extremidad |
| Grado 2. Movimiento realizado sin gravedad con todo o más de la mitad del rango de movimiento |
| Grado 3. Movimiento contra la gravedad en todo o más de la mitad del rango de movimiento |
| Grado 4. Movimiento contra resistencia leve-moderada en todo el rango de movimiento |
| Grado 5. Potencia de contracción normal (resistencia fuerte) |
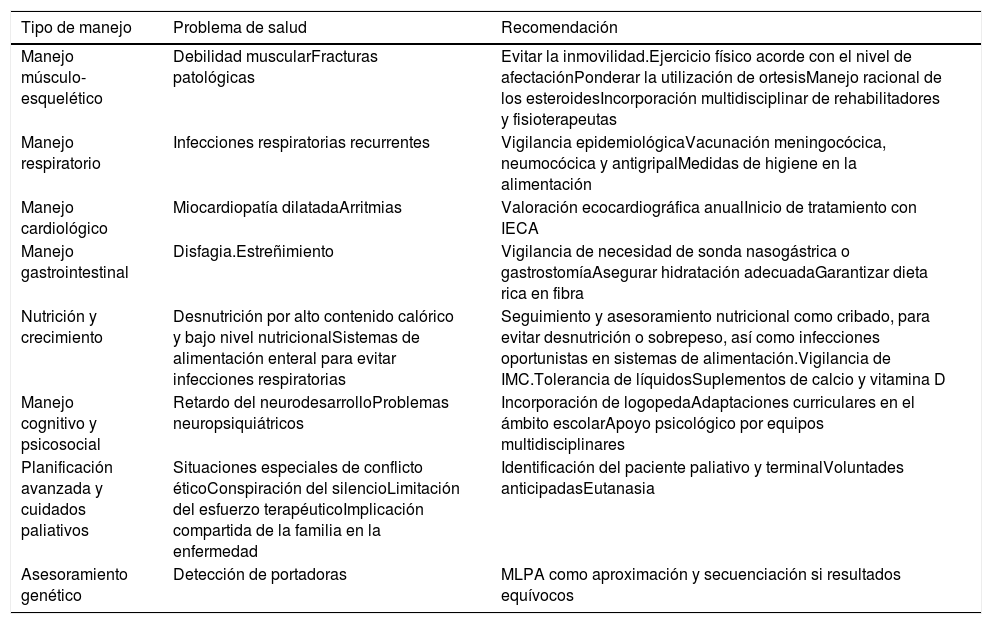
Tratamientos no farmacológicos en DMD
| Tipo de manejo | Problema de salud | Recomendación |
|---|---|---|
| Manejo músculo-esquelético | Debilidad muscularFracturas patológicas | Evitar la inmovilidad.Ejercicio físico acorde con el nivel de afectaciónPonderar la utilización de ortesisManejo racional de los esteroidesIncorporación multidisciplinar de rehabilitadores y fisioterapeutas |
| Manejo respiratorio | Infecciones respiratorias recurrentes | Vigilancia epidemiológicaVacunación meningocócica, neumocócica y antigripalMedidas de higiene en la alimentación |
| Manejo cardiológico | Miocardiopatía dilatadaArritmias | Valoración ecocardiográfica anualInicio de tratamiento con IECA |
| Manejo gastrointestinal | Disfagia.Estreñimiento | Vigilancia de necesidad de sonda nasogástrica o gastrostomíaAsegurar hidratación adecuadaGarantizar dieta rica en fibra |
| Nutrición y crecimiento | Desnutrición por alto contenido calórico y bajo nivel nutricionalSistemas de alimentación enteral para evitar infecciones respiratorias | Seguimiento y asesoramiento nutricional como cribado, para evitar desnutrición o sobrepeso, así como infecciones oportunistas en sistemas de alimentación.Vigilancia de IMC.Tolerancia de líquidosSuplementos de calcio y vitamina D |
| Manejo cognitivo y psicosocial | Retardo del neurodesarrolloProblemas neuropsiquiátricos | Incorporación de logopedaAdaptaciones curriculares en el ámbito escolarApoyo psicológico por equipos multidisciplinares |
| Planificación avanzada y cuidados paliativos | Situaciones especiales de conflicto éticoConspiración del silencioLimitación del esfuerzo terapéuticoImplicación compartida de la familia en la enfermedad | Identificación del paciente paliativo y terminalVoluntades anticipadasEutanasia |
| Asesoramiento genético | Detección de portadoras | MLPA como aproximación y secuenciación si resultados equívocos |
Manejo respiratorio: para una correcta evolución se contempla la incorporación de un neumólogo y un terapeuta especializado en fisioterapia respiratoria. Teniendo en cuenta el alto riesgo de infecciones respiratorias, debe incluirse una inmunización frente a patógenos como el neumococo y la gripe4,5,8.
Manejo cardiológico: el papel del cardiólogo es evitar clínica de insuficiencia cardíaca y el tratamiento de las arritmias acompañantes mediante diagnóstico precoz de la miocardiopatía dilatada primaria. El uso de inhibidores de la enzima conversora de angiotensina es la opción terapéutica de primera línea. El inicio de la terapia con agentes antiarrítmicos se utiliza para el tratamiento de la fibrilación auricular y las arritmias ventriculares5,8,16.
Manejo gastrointestinal: es necesario un seguimiento nutricional periódico y ajustes en la dieta. En fases avanzadas aparece disfagia, en este caso se puede considerar la alimentación por sonda nasogástrica y botón gástrico. Los pacientes también presentan reflujo gastroesofágico y esofagitis por lo que es importante el uso de inhibidores de la bomba de protones o antagonistas del receptor H2. También se debe estar atento al estreñimiento para comenzar tratamiento específico, asegurar una buena hidratación y una ingesta adecuada5,11.
Nutrición y crecimiento: el profesional de AP puede ser un excelente recurso para el seguimiento y el asesoramiento nutricional general a lo largo del tiempo. En la DMD, ha de prestarse especial atención para asegurar una ingesta adecuada de líquidos, de calcio y vitamina D, y evitar alimentos con mal perfil nutricional (con alto contenido calórico, pero bajo contenido nutricional [p.-ej., comida rápida]). El papel del nutricionista en asesoramiento y seguimiento, es muy aconsejable15,17,18.
Obtener unas medidas de peso y altura en estos pacientes para un control adecuado del crecimiento es difícil, especialmente en formas avanzadas donde los pacientes, ya en la adolescencia, van en silla de ruedas. Por tanto, la medida del índice de masa corporal puede ser poco fiable.
La disfagia condiciona la alimentación oral, por lo que puede ser preciso sonda nasogástrica y, en periodos avanzados, gastrostomía, para evitar las infecciones pulmonares recurrentes y garantizar un adecuado aporte calórico16. No es necesario alcanzar un peso que se considere saludable para pacientes no afectados de la misma edad y sexo, ya que el exceso de peso puede ser perjudicial para la movilización del paciente y las complicaciones respiratorias5,17.
Manejo cognitivo y psicosocial: es frecuente la aparición de retraso del neurodesarrollo en la infancia temprana, problemas cognitivos y del aprendizaje y problemas neuropsiquiátricos, pero es frecuente que no tengan el manejo adecuado. Es necesario valorar la necesidad de logopeda, psicoterapia o adaptación curricular a nivel escolar para el desarrollo del niño con DMD2,4,5.
Planificación avanzada y cuidados paliativos: cuando las circunstancias lo permitan, los profesionales sanitarios deben involucrar a los pacientes y sus familias en las discusiones sobre las opciones de tratamiento, la planificación de la atención avanzada, las voluntades anticipadas, los cuidados paliativos y otros temas relacionados con el final de la vida, según los valores ético-morales y preferencias del paciente5,19.
Del mismo modo, la atención educativa ha de estar bien centrada en las condiciones patológicas del paciente, por tanto, es importante que profesionales de los diferentes niveles educativos estén involucrados con la familia y los pacientes para un manejo diario y la detección precoz de complicaciones19.
Asesoramiento genético: para el asesoramiento genético es imprescindible detectar a las madres portadoras de una mutación. Es importante ofrecer estos estudios a las mujeres con familiares enfermos y que tengan deseo gestacional5,20.
Actualización para el tratamiento de la distrofia muscular de DuchenneActualmente hay 2fármacos aprobados como terapia específica para la DMD. Se basan en la administración de moléculas, nucleósidos o no nucleósidos, que permitan una lectura genética adecuada con supresión de las mutaciones y oligonucleótidos antisentido que permiten la omisión del exón enfermo3,21. Atalureno se utiliza para mutaciones sin sentido en el gen de la distrofina, lo que da lugar a la transformación de una proteína truncada no funcional en otra funcional. Se utiliza en pacientes ambulantes a partir de 2 o más años de edad12. Eteplirsen es un oligonucleótido antisentido de síntesis diseñado para saltar el exón 51, permitiendo recuperar la pauta de lectura en algunas deleciones específicas de la región central del gen11,21,22.
Ha sido publicado recientemente un metaanálisis sobre el atalureno donde se realizan 2ensayos, uno en fase iib (ClinicalTrials.gov: NCT00592553) y el ACT DMD, en fase iii. En total se recluta y se analiza a 342 pacientes, 171 en la rama de atalureno y 171 en la rama placebo, durante 48 semanas. Se detectó que existe un beneficio estadísticamente significativo a favor de atalureno en el test de la marcha de los 6 min, en especial en el análisis por subgrupos de los enfermos con test de la marcha a 6 min entre 300 y 400 m (en la zona de transición entre bajo y riesgo alto de progresión de enfermedad) y en aquellos que no alcanzaban los 300 m23.
El ensayo 202 sobre eteplirsen estudia la evolución de 12 pacientes del estudio 201 (test de la marcha a los 6 min entre 200 y 400 m) con antecedente de mutación confirmada genéticamente en el exón 51 en tratamiento con corticoides y eteplirsen a largo plazo24. De los 12 pacientes, 2 hermanos gemelos idénticos experimentaron una pérdida rápida y temprana de la deambulación, los otros 10 pacientes mantuvieron la capacidad de marcha. Los gemelos tenían una mayor gravedad de la enfermedad al inicio del estudio. En el trabajo se comparan la función cardíaca, función pulmonar y fuerza de las extremidades superiores, y la producción de distrofina en los pacientes gemelos no deambulantes versus los 10 pacientes ambulantes durante 240 semanas de tratamiento combinado. Los pacientes más graves mantuvieron la función cardíaca y la fuerza de las extremidades superiores durante el tiempo de tratamiento en rangos similares a las formas menos graves. La producción de distrofina aumentó en los grupos con eteplirsen en comparación con los pacientes que no reciben este tratamiento, incluyendo a los pacientes con enfermedad más avanzada. A pesar de la pérdida de la deambulación, otros marcadores de progresión de la enfermedad se mantuvieron relativamente estables en los pacientes gemelos tratados con eteplirsen y fueron similares a los de los pacientes ambulatorios25.
En investigación hay distintos tipos de proyectos, si bien ninguno de ellos está focalizado en el ámbito de la AP:
- –
La fisiopatología de la DMD también se caracteriza por una síntesis alterada de óxido nítrico debido a una reducción de la actividad de la sintasa de dicho óxido nítrico, provocando oxidación, inflamación y estrés metabólico. Está en estudio el papel de fármacos donantes de óxido nítrico (como dinitrato de isosorbida) junto a antiinflamatorios no esteroideos3,5,25. Además se están ensayando fármacos para el tratamiento de la fibrosis muscular (tamoxifeno o coenzima Q10)3,21. En la preservación de la función pulmonar ya hay resultados sobre idebenona, que en el estudio DELOS parece conservar dicha función pulmonar sin el uso de corticoide, incluso en el seguimiento a largo plazo parece mantener sus ventajas en la capacidad vital y PEF (peak expiratory flow percent of predicted), con las posibles ventajas a nivel de complicaciones respiratorias26,27. En desarrollo están los ensayos sobre el crecimiento muscular y regeneración muscular (domagrozumab y talditercept alfa)28, así como en la esfera cardiológica para disminuir el impacto de la miocardiopatía con fármacos de uso conocido, tales como ramipril, carvedilol y nevibolol3,16. En un estudio aleatorizado reciente sobre el uso de enalapril y metoprolol en el inicio de la disfunción ventricular izquierda27 se detecta que el uso de estos tratamientos puede retrasar la progresión de la miocardiopatía intrínseca a insuficiencia cardíaca ventricular izquierda (muestra de 38 pacientes entre 10 y 14 años con fracción de eyección de ventrículo izquierdo mayor de 30% al inicio del estudio)28.
- –
Terapias genéticas: su fundamento es introducir el gen sano de la distrofina a través de un vector a las células de músculo esquelético y cardíaco con el fin de restaurar la proteína de la distrofina. Debido al gran tamaño del gen se están intentando infusiones de minidistrofina en animales de laboratorio. Una de las dificultades mayores es la respuesta inmune del huésped buscando, en los nuevos estudios, vectores quiméricos29,30.
- –
Terapia celular: ofrecen la oportunidad de reemplazar la distrofina enferma como cura potencial. Para ello hay en marcha distintos ensayos clínicos con células madre29,30. Como ejemplo reciente, el ensayo clínico HOPE Duchenne16 analiza 25 pacientes mayores de 12 años con fibrosis miocárdica. Doce pacientes con cuidados habituales y 13 enfermos son tratados con terapia celular intracoronaria (CAP-1002). Tras 12 meses de seguimiento parece que CAP-1002 intracoronario en DMD es seguro y demuestra señales de eficacia en función cardíaca y de las extremidades superiores.
- –
El papel de la utrofina y su modulación. Esta línea se basa en que la utrofina es el homólogo de la distrofina y en el músculo adulto se localiza en las uniones neuromusculares y miotendinosas. La expresión de utrofina aumenta en los pacientes con DMD debido a la ausencia de distrofina22,29. Se piensa que la utrofina puede actuar como un sustituto eficaz de la distrofina en los músculos; el ezutromid está aportando resultados muy interesantes30-32.
- –
El uso de nanotecnología como sistema de administración no viral de fármacos y macromoléculas, como ADN plasmídico, oligonucleótidos y proteínas, a través de varias vías de administración. Es una de las herramientas esperanzadoras para que las terapias anteriormente descritas puedan llegar de forma selectiva a su diana para realizar un tratamiento selectivo y eficaz31,32. De hecho, existen actualmente, varios ensayos clínicos en distintas fases de experimentación, algunos ya pendientes de los últimos flecos legales para ser aprobados para su administración31.
El papel del médico especialista en AP y domiciliaria es fundamental dado que va a ser el médico que brinde la asistencia continuada y más cercana al paciente y su entorno. Ahora se detallarán algunas recomendaciones específicas para estos especialistas, sin perder la perspectiva del trabajo multidisciplinar y la interrelación entre el resto de especialistas hospitalarios y la AP (tabla 5)2,6.
- 1.
Inmunización: se debe cumplir el calendario vacunal vigente en el niño con DMD, en especial aquellas con virus no vivos. En el caso de que se decida administrar virus vivos atenuados, se debe realizar antes de comenzar el tratamiento corticoideo; estar vacunas están contraindicadas en el paciente con DMD y dosis alta de corticoides (dosis> 20mg o 2mg/kg/día de prednisona o equivalente). Es recomendable la vacunación antigripal anual del paciente y de sus contactos estrechos. Por último, también es recomendable la vacuna antineumocócica del paciente con DMD por el riesgo de complicaciones respiratorias2,6.
- 2.
Nutrición: brindar una adecuada educación nutricional para evitar complicaciones como la malnutrición o la obesidad. Planificar una dieta saludable, teniendo en cuenta en los pacientes con tratamiento con corticoides la ingesta de calcio y vitamina D. Se debe tener un plan de seguimiento en consulta, tanto de la nutrición como del crecimiento. Por otro lado, en relación con la nutrición, no olvidar la salud bucodental y hacer un seguimiento de la misma de forma periódica (sobre todo en los pacientes con bifosfonatos por osteoporosis secundaria a corticoides)5,15,17,33.
- 3.
Seguridad: es necesario explicar a los familiares y al propio paciente, si es posible, el riesgo de caídas y accidentes en las actividades cotidianas. Se debe usar ortesis y medios de transporte adaptados y adecuados, por ejemplo, valorar si dispositivos, como la silla de ruedas, cumplen los requisitos de seguridad en caso de no tener un control adecuado del tronco. La atención urgente ha de estar integrada e interrelacionada con la AP, existiendo modelos de atención compartida como ocurre en otras enfermedades neuromusculares como la esclerosis lateral amiotrófica2,4.
- 4.
Vigilancia de complicaciones: cuando un paciente acude a revisión se debe atender tanto a las complicaciones propias de la enfermedad como a las derivadas de los tratamientos. Respecto a la enfermedad, se deben valorar complicaciones como traumatismos o riesgos de caídas, enfermedad respiratoria, riesgo de broncoaspiraciones o complicaciones digestivas/disfagias, riesgo de cardiopatía y signos de insuficiencia cardíaca, alteraciones nutricionales y del crecimiento y, finalmente, no se debe olvidar las alteraciones del manejo cognitivo y psicosocial desde la visión de la AP. Por otro lado, en las complicaciones medicamentosas se debe tener en cuenta el uso de terapias específicas y el uso de esteroides (que cada vez es más precoz), vigilando en este caso la aparición de insuficiencia suprarrenal, osteoporosis, obesidad, alteración de glucemia capilar o hipertensión entre otros efectos secundarios. Los anestésicos por vía intravenosa están indicados frente a los inhalados por el riesgo de rabdomiólisis e hipertermia maligna. Existe contraindicación formal sobre los relajantes musculares despolarizantes2,12,13.
- 5.
Atención continuada: el seguimiento periódico del paciente va a ser la forma de poder anticiparnos a las distintas complicaciones que se pueden presentar. Es imprescindible la educación sanitaria en el paciente y sus familiares, facilitando el acceso a una información veraz y en el empoderamiento de su enfermedad, permitiendo así conocer el tratamiento y el pronóstico. Evitar el aislamiento social de los pacientes por su enfermedad tanto de la familia, compañeros escolares como de la comunidad en general. Fomentar el acceso al movimiento asociativo correspondiente, especialmente en aquellos lugares donde esté desarrollado. Dado que los pacientes tienen edad pediátrica, sería muy interesante enfocar la enfermedad desde el punto de vista escolar teniendo acceso a la salud escolar y formando a los maestros y profesores sobre el comportamiento en las aulas y los signos de alarma cuando estén bajo su mando1,19,29.
- 6.
Atención al final de la vida: es necesario tener una comunicación de futuro, con la información correcta y con la confianza para poder planificar las actuaciones. La AP puede ser una especialidad ideal en este sentido, por el acompañamiento durante todo el proceso y la posibilidad de conocer con detalle las circunstancias que acompañan al enfermo. En caso de haber dado una opinión, se debe respetar siempre la voluntad del paciente y ofrecer las medidas disponibles para cada etapa. Finalmente, siempre tenemos que contar con la opinión de la familia y acompañarlos con especial atención en las etapas finales de esta patología. La coordinación con los equipos de cuidados paliativos es fundamental, tanto para la identificación del paciente paliativo como para aquellos pacientes en situación de terminalidad. Hay que estar atento a los documentos de voluntades vitales anticipadas, así como a los deseos de eutanasia que los pacientes pueden solicitar.
- 7.
Prevención: el asesoramiento genético es clave para la detección de las mujeres portadoras. Existirán mujeres que querrán evitar el deseo genésico y habrá que fomentar la terapia anticonceptiva cuando sea preciso y demandado. Fomentar el acceso a la información de genética aquellos pacientes y familiares que lo soliciten, empleando un lenguaje útil y accesible11,12.
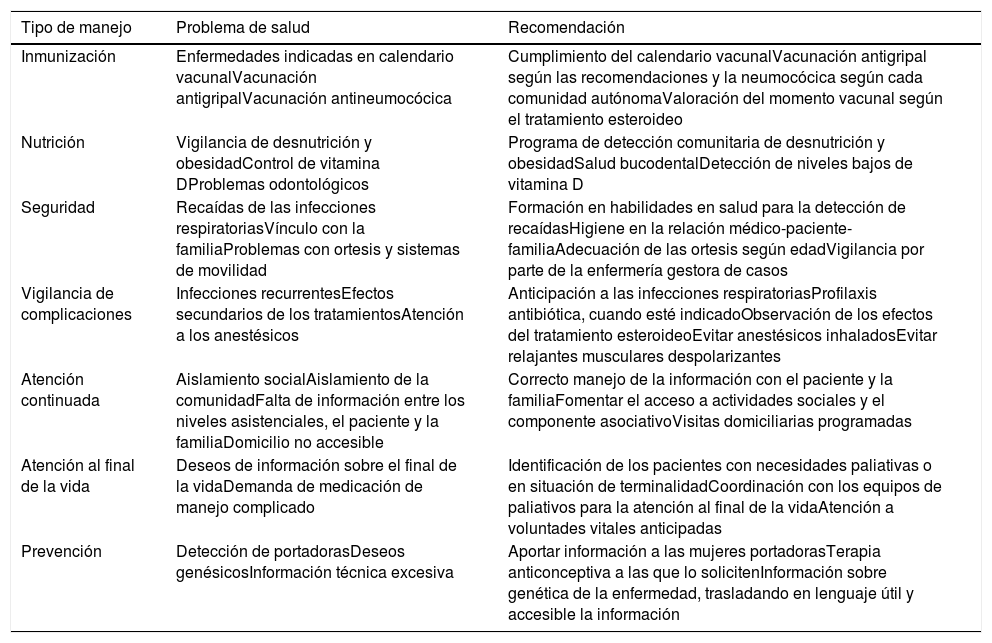
Tipo de recomendaciones desde Atención Primaria
| Tipo de manejo | Problema de salud | Recomendación |
|---|---|---|
| Inmunización | Enfermedades indicadas en calendario vacunalVacunación antigripalVacunación antineumocócica | Cumplimiento del calendario vacunalVacunación antigripal según las recomendaciones y la neumocócica según cada comunidad autónomaValoración del momento vacunal según el tratamiento esteroideo |
| Nutrición | Vigilancia de desnutrición y obesidadControl de vitamina DProblemas odontológicos | Programa de detección comunitaria de desnutrición y obesidadSalud bucodentalDetección de niveles bajos de vitamina D |
| Seguridad | Recaídas de las infecciones respiratoriasVínculo con la familiaProblemas con ortesis y sistemas de movilidad | Formación en habilidades en salud para la detección de recaídasHigiene en la relación médico-paciente-familiaAdecuación de las ortesis según edadVigilancia por parte de la enfermería gestora de casos |
| Vigilancia de complicaciones | Infecciones recurrentesEfectos secundarios de los tratamientosAtención a los anestésicos | Anticipación a las infecciones respiratoriasProfilaxis antibiótica, cuando esté indicadoObservación de los efectos del tratamiento esteroideoEvitar anestésicos inhaladosEvitar relajantes musculares despolarizantes |
| Atención continuada | Aislamiento socialAislamiento de la comunidadFalta de información entre los niveles asistenciales, el paciente y la familiaDomicilio no accesible | Correcto manejo de la información con el paciente y la familiaFomentar el acceso a actividades sociales y el componente asociativoVisitas domiciliarias programadas |
| Atención al final de la vida | Deseos de información sobre el final de la vidaDemanda de medicación de manejo complicado | Identificación de los pacientes con necesidades paliativas o en situación de terminalidadCoordinación con los equipos de paliativos para la atención al final de la vidaAtención a voluntades vitales anticipadas |
| Prevención | Detección de portadorasDeseos genésicosInformación técnica excesiva | Aportar información a las mujeres portadorasTerapia anticonceptiva a las que lo solicitenInformación sobre genética de la enfermedad, trasladando en lenguaje útil y accesible la información |
- 1.
La DMD (DM1) es una miopatía con afectación multiorgánica que se diagnostica, generalmente, en la infancia.
- 2.
Las mujeres son portadoras y no presentan la enfermedad.
- 3.
Las infecciones respiratorias son la primera causa de mortalidad en la DMD, tanto por la debilidad muscular como por los efectos de inmunodepresión por el tratamiento esteroideo, que cada vez ha de ser más precoz.
- 4.
Esa debilidad muscular genera problemas digestivos (disfagia) e infecciones respiratorias recurrentes.
- 5.
Los pacientes necesitan apoyo psicosocial, no solo de profesionales sanitarios, sino de la red comunitaria y del movimiento asociativo.
- 6.
La coordinación entre niveles asistenciales AP y Urgencias, AP y Atención Hospitalaria, y AP y Equipo de Cuidados Paliativos ha de ser reforzada, no solo en la DMD, sino en enfermedades invalidantes, especialmente en un mundo sanitario con cada vez más proyección no presencial.
- 7.
El aumento de la esperanza de vida de estos pacientes hace que los enfermos con DMD sean pacientes de AP, por lo que los MAP han de tener un papel de relevancia por la transversalidad de esta especialidad y por la continuidad asistencial del paciente durante toda su vida.
Los autores declaran no tener ningún conflicto de intereses, ni han recibido financiación de ningún tipo para el desarrollo de este trabajo.
