




Los estudios de investigación con seres humanos, sus muestras biológicas o sus datos de carácter personal en el ámbito de la biomedicina han sido ya desde mediados del siglo pasado objeto de regulación. Inicialmente una regulación basada en recomendaciones como el Código de Nuremberg, el Informe Belmont o las primeras versiones de la Declaración de Helsinki. Todos ellos documentos en los que fueron conceptualizando los principios (autonomía, beneficencia, no-maleficencia y justicia) que todo investigador había de seguir en el desarrollo de su investigación. Esta primera fase se conoce como período de autorregulación porque se considera que los propios investigadores podrían, siguiendo esas recomendaciones, llevar a cabo sus investigaciones sin otro control. Posteriormente, se pasó a un período claramente regulatorio en el que las premisas de estas recomendaciones fueron progresivamente incorporándose al ordenamiento jurídico de los distintos países y con ello surgió el control externo de la investigación por parte de las administraciones y de otros órganos como los Comités de Ética de la Investigación.
La finalidad de este artículo es servir como guía a los profesionales cuya actividad principal es la asistencial en el ámbito de la atención primaria y que a su vez están interesados en iniciar estudios de investigación para responder a las incertidumbres que en el contexto de su actividad diaria les puedan surgir.
Studies of research with human beings, their biological specimens, or their personal data in the field of biomedicine have been subject to regulation since the middle of the last century. Initially a regulation based on recommendations such as the Nuremberg Code, the Belmont Report or the first versions of the Declaration of Helsinki. All of them documents in which the principles (autonomy, beneficence, non-maleficence, and justice) were conceptualized, and that all researchers had to follow in the development of their research. This first phase is known as a period of self-regulation, because it is considered that the researchers themselves could, by following these recommendations, carry out their investigations without further control. Subsequently, it went through a clearly regulatory period in which the premises of these recommendations were progressively incorporated into the legal system of the different countries, and with this, arose the external control of the investigation by the administrations and other bodies, such as the Research Ethics Committees.
The purpose of this article is to serve as a guide to professionals whose main activity is care in the field of Primary Care and who, in turn, are interested in initiating research studies to respond to uncertainties in the context of their daily activity that may arise.
Los estudios de investigación con seres humanos, sus muestras biológicas o sus datos de carácter personal en el ámbito de la biomedicina han sido ya desde mediados del siglo pasado objeto de regulación. Inicialmente una regulación basada en recomendaciones como el Código de Nuremberg1, el Informe Belmont2 o las primeras versiones de la Declaración de Helsinki3.
Todos ellos documentos en los que fueron conceptualizando los principios (autonomía, beneficencia, no-maleficencia y justicia) que todo investigador había de seguir en el desarrollo de su investigación. Esta primera fase se conoce como período de autorregulación porque se considera que los propios investigadores podrían, siguiendo esas recomendaciones, llevar a cabo sus investigaciones sin otro control. Posteriormente, se pasó a un período claramente regulatorio en el que las premisas de estas recomendaciones fueron progresivamente incorporándose al ordenamiento jurídico de los distintos países y con ello surgió el control externo de la investigación por parte de las administraciones y de otros órganos como los Comités de Ética de la Investigación.
La finalidad de este artículo es servir como guía a los profesionales cuya actividad principal es la asistencial en el ámbito de la atención primaria y que a su vez están interesados en iniciar estudios de investigación para responder a las incertidumbres que en el contexto de su actividad diaria les puedan surgir4,5.
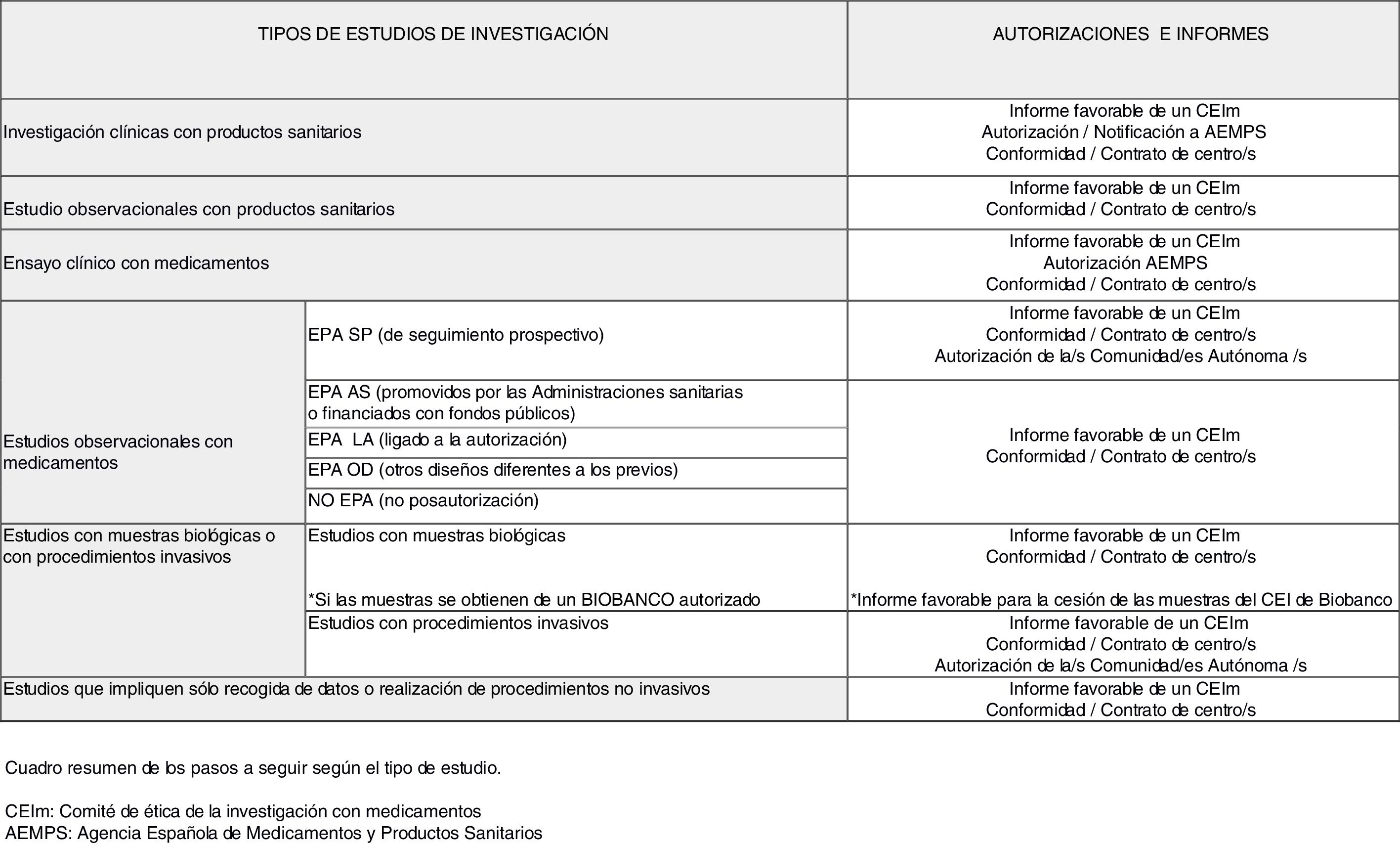
Pasos para poner en marcha un estudio de investigación biomédicaA continuación, se ha intentado reproducir en orden las preguntas que, una vez tengamos claro los objetivos del estudio que se quiere iniciar, se debe hacer el investigador para conocer los pasos a seguir con el fin de obtener todos los dictámenes, autorizaciones o conformidades que sean precisos antes del inicio del estudio. En la figura 1 se ha intentado resumir el complejo campo normativo de la investigación biomédica.

Así, teniendo en cuenta la legislación vigente hoy en día en España, la primera pregunta que tendría que hacerse es:
1. ¿Implica el estudio medicamentos?
En caso afirmativo, debemos plantearnos 2 opciones: ¿se trata de un estudio observacional o de un estudio experimental?
La principal diferencia entre ambos estudios está en el papel del investigador. Por una parte, en un estudio observacional el investigador observa lo que ocurre en la práctica habitual y lo único que diferenciará su futura actividad en el estudio de la práctica habitual será que adicionalmente a esta utilizaremos tiempo en recoger datos clínicos para analizarlos; aquí se encuadran los estudios tipo registros o estudios de cohortes que son los más habituales en atención primaria6. Sin embargo, en un estudio experimental (ensayo clínico) el investigador interviene directamente sobre los sujetos de investigación7 de la siguiente forma: a) asignando a los sujetos medicamentos no autorizados o en indicaciones no contempladas en su ficha técnica (quizás los 2 tipos que más frecuentemente se relacionan con el concepto de ensayo clínico con medicamentos), b) cuando toma la decisión de prescribir los medicamentos en investigación junto con la de incluir al sujeto en el estudio, o la deja al azar y c) cuando utiliza procedimientos de diagnóstico o seguimiento que no se usarían en la práctica habitual.
1.1 Estudios observacionales con medicamentos
Si nuestro estudio entra en la definición de observacional en la actualidad viene regulado por la Orden SAS/3470/009, de 16 de diciembre, por la que se publican las directrices sobre estudios postautorización de tipo observacional para medicamentos de uso humano8. En esta orden se establece que todos los estudios que se realicen con seres humanos o con registros médicos y que tengan uno o varios medicamentos como exposición de interés deben ser remitidos a la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) para que esta emita una resolución de los mismos indicando la clasificación del estudio de acuerdo a unos tipos definidos en la mencionada orden. Dependiendo de en qué tipo sea clasificado, las autorizaciones y dictámenes que se han de obtener antes del inicio son diferentes:
1.1.a Estudio observacional de seguimiento prospectivo (EPA SP): Es decir, aquellos estudios de carácter observacional (en el sentido que se describió previamente), en los que tras la selección inicial de los pacientes, por tener un problema de salud o por estar a tratamiento con un medicamento, se plantea un seguimiento prospectivo durante un tiempo definido en el protocolo para la recogida de los datos clínicos que se consideren de interés (variables del estudio) para responder a las hipótesis planteadas.
En caso de que el estudio sea clasificado como EPA SP es preciso en primer lugar presentarlo para evaluación a un Comité de Ética de la Investigación con medicamentos (CEIm). Los CEIm son comités de ética de la investigación (CEI) que están acreditados para emitir un dictamen en un estudio clínico con medicamentos y en una investigación clínica con productos sanitarios9. El listado de CEIm acreditados en España está disponible en la página web de la AEMPS, en el apartado de investigación clínica con medicamentos.
Una vez obtenido el dictamen favorable de un CEIm es preciso que sea autorizado por cada una de las comunidades autónomas donde se va a realizar. Esta autorización administrativa viene motivada porque son estudios que han sido tradicionalmente utilizados por la industria farmacéutica con el objetivo de promocionar los medicamentos que se han comercializados en fecha reciente, ya que pueden fomentar su prescripción (son los denominados estudios de siembra, seeding trials o marketing trials).
Para conocer la unidad administrativa que se encarga de gestionar estas autorizaciones en cada comunidad autónoma, se ha de consultar también en este caso la página web de la AEMPS. Por desgracia, en este caso, y hasta que la citada Orden SAS/3470/009 no sea modificada, cada comunidad autónoma tiene sus propios requisitos para obtener la citada autorización.
Por último, una vez obtenidos el dictamen favorable de un CEIm y la autorización de cada una de las comunidades autónomas donde al menos haya un centro en el que se quiera llevar a cabo el estudio, se ha de contactar con los centros para informarles del mismo entregándoles copia del protocolo, dictamen del CEIm y la autorización de la comunidad autónoma. En la práctica, hay centros que simplemente emitirán un documento de conformidad al inicio del estudio en sus instalaciones (en especial, en aquellos EPA SP que no tengan asociada una memoria económica en la que se prevea remuneración a los investigadores) mientras que habrá otros centros que exijan la firma de un contrato entre las partes (centro sanitario, promotor e investigador del estudio).
1.1.b Estudio observacional promovidos por las administraciones sanitarias o financiados con fondos públicos (EPA AS): Es decir, aquellos estudios de carácter observacional (en el sentido que se describió previamente), que han sido financiados por el Instituto de Salud Carlos III (ISCIII), Ministerio de Economía y Competitividad, convocatorias europeas…, o estudios que promueven directamente la administración sanitaria, por ejemplo las consejerías de salud de las comunidades autónomas.
En este caso y con la finalidad de facilitar su realización no se exige la autorización a cada una de las comunidades autónomas donde se va a realizar, sino que solo será remitido al Comité de Coordinación de Estudios Postautorización (comité ligado a la AEMPS). Seguirá siendo necesario el dictamen favorable de un CEIm y de nuevo se ha de contactar con los centros para informarles del mismo entregándoles copia del protocolo, dictamen del CEIm y la resolución de la AEMPS. El hecho de que se requiera o no la firma de un contrato entre las partes de nuevo queda a discreción de cada centro.
1.1.c Estudio observacional con otros diseños (EPA OD): Es decir, aquellos estudios de carácter observacional (en el sentido que se describió previamente), en los que tras la selección inicial de los pacientes por tener un problema de salud o por estar a tratamiento con un medicamento, no se plantea un seguimiento prospectivo. Son, por ejemplo, los estudios transversales o los estudios en los que se escoge un diseño tipo casos y controles.
En este caso es preciso el dictamen favorable de un CEIm, así como realizar el mismo trámite que en los 2 casos anteriores para la obtención de las conformidades de los centros donde se vaya a iniciar.
1.1.d Estudio observacional no postautorización (No EPA): Son estudios observacionales en los que, si bien se pueden recoger datos de medicamentos, el factor de exposición fundamental investigado no son estos (por ejemplo, medir el impacto de las medidas dietéticas y el estilo de vida saludable en los pacientes diabéticos en el control metabólico de la enfermedad). La ruta a seguir coincidiría con la previa.
En todos estos estudios la orden que los regula es claro e indica que con el dictamen favorable de un único CEIm acreditado en España es suficiente. Sin embargo, en la práctica, hoy en día, muchos centros siguen condicionando la emisión de su conformidad para que se inicien en su ámbito a que sean evaluados o presentados (al menos) al CEIm que los tutela. Por lo tanto, la recomendación es que en el caso de que el estudio en cuestión sea multicéntrico, se presente inicialmente a un CEIm (aquel que el promotor/investigador coordinador seleccione) y se obtenga el dictamen favorable. Una vez obtenido este dictamen favorable, este se remita junto con los documentos definitivos aprobados por ese comité (protocolo y documentos de consentimiento informado, y memoria económica si hubiera) al resto de los centros. También sería recomendable como fase previa hacer un análisis de los requisitos que exigen en cada uno de los centros en los que se prevea realizar para valorar los costes (no solo económicos sino también en tiempo y esfuerzo del equipo investigador) y por tanto la viabilidad del estudio. En esta fase también se pueden identificar aquellos centros que no condicionan la emisión de su conformidad para que se inicien en su ámbito a que sean evaluados por el CEIm que los tutela, y así centrar los esfuerzos (en caso de recursos limitados) en estos.
La citada orden también regula otros estudios postautorización de tipo observacional, los estudios EPA LA (ligados a la autorización) que son una condición establecida en el momento de la autorización de un medicamento al titular de la comercialización, o una exigencia de la autoridad competente para aclarar cuestiones relativas a la seguridad del medicamento o forman parte del plan de gestión de riesgos. No se detallan los procedimientos aplicables a estos estudios, pues dadas sus características se entiende que no serían estudios promovidos por un investigador o grupo de investigadores independientes de la industria farmacéutica.
1.2. Ensayos clínicos con medicamentos (estudios experimentales)
Habitualmente relacionamos ensayos clínicos con medicamentos con estudios en los que se prueban medicamentos aun no autorizados por las autoridades regulatorias. Es cierto que estos son un alto porcentaje de los ensayos clínicos que se realizan en nuestro país y en otros. Sin embargo, de acuerdo con el real decreto que los regula en España9 y con el Reglamento europeo de ensayos clínicos10, entraría en la definición de ensayo clínico con medicamento todo estudio en el que:
- a)
se asigna de antemano al sujeto de ensayo a una estrategia terapéutica determinada, que no forma parte de la práctica clínica habitual del Estado miembro implicado;
- b)
la decisión de prescribir los medicamentos en investigación se toma junto con la de incluir al sujeto en el estudio clínico;
- c)
se aplican procedimientos de diagnóstico o seguimiento a los sujetos de ensayo que van más allá de la práctica clínica habitual.
Es decir, se trataría de un ensayo clínico cuando en nuestro estudio usamos un medicamento para una indicación en la que no se emplea en la práctica clínica habitual.
También sería un ensayo clínico cuando, si bien los medicamentos los empleamos siguiendo las condiciones de la práctica habitual, la decisión de esta prescripción la dejamos al azar (es decir, por ejemplo, aleatorizamos a un grupo de pacientes con dolor a 2 AINE diferentes, ibuprofeno y dexketoprofeno). O cuando en nuestro estudio se usa un medicamento siguiendo las condiciones de la ficha técnica pero realizamos procedimientos distintos a la práctica habitual (por ejemplo, estudio para evaluar la eficacia del tratamiento antiagregante/anticoagulante en los cambios en la placa de ateroma en pacientes con cardiopatía isquémica en los que se plantea la realización de cateterismos adicionales al estándar recomendado en las guías para la obtención de muestras y evaluación de la variable principal).
Los estudios que entren en esta categoría deberán seguir el procedimiento previsto en el Real Decreto 1090/2015, de 4 de diciembre, por el que se regulan los ensayos clínicos con medicamentos, los CEIm y el Registro Español de Estudios Clínicos (https://reec.aemps.es/reec/public/web.html). En este caso, antes del inicio del ensayo en los centros se ha de obtener el dictamen de un CEIm acreditado, la autorización de la AEMPS, así como la conformidad del centro. Desde la entrada en vigor del decreto en 2016, el promotor del estudio debe remitir simultáneamente a la AEMPS y al CEIm que elija a través del portal de ensayos clínicos (https://sede.aemps.gob.es/usoHum/ensaClin/portal_ensaClinicos.htm#documentacion) la documentación prevista en el Memorando de Colaboración e Intercambio de Información entre la AEMPS y los CEIm11. La conformidad de los centros de nuevo podrá expresarse a través de la firma de un contrato entre las partes (promotor, centro e investigador). En aquellos casos en los que el promotor sea un profesional del centro y no se requiera firma de contrato se precisará la conformidad expresa de la dirección del centro participante. Como en los estudios observacionales con medicamentos, en esta situación de nuevo queda a discreción de cada centro el elegir la fórmula de cómo expresar esta conformidad.
Un aspecto importante que reguló el Real Decreto 1090/20159, y que puede ser de especial interés para los investigadores independientes de la industria farmacéutica que a menudo no cuenta con una fuente de financiación o esta es escasa, es la figura de ensayo clínico de bajo nivel de intervención. Este tipo de ensayo clínico está extendiéndose mucho en atención primaria, siendo este ámbito el principal campo de desarrollo de los ensayos de bajo nivel de intervención.
Esta definición se refiere a ensayos clínicos en los que todos los medicamentos en investigación utilizados están autorizados y se utilizan de conformidad con los términos de la autorización de comercialización, o bien su uso no está contemplado en la ficha técnica, pero se basa en pruebas y está respaldado por datos científicos publicados para esa indicación. Además, todos los procedimientos complementarios de diagnóstico o seguimiento que se llevan a cabo siguiendo el protocolo del ensayo implican un riesgo o carga adicional para la seguridad de los sujetos que es mínimo comparado con el de la práctica clínica habitual en esa población. Debe ser el promotor del ensayo el que solicite la clasificación como bajo nivel de intervención para lo cual lo ha de justificar, y será el CEIm y la AEMPS quien tras la evaluación lo acepten o no. En la resolución de autorización de la AEMPS se dejará constancia de que el ensayo ha sido clasificado como tal. Es muy importante tener en cuenta que para los ensayos clínicos de bajo nivel de intervención, tal como contempla el mencionado decreto, no sería preciso contratar un seguro específico que cubra los daños y perjuicios que pudieran sufrir los participantes, siempre y cuando las pólizas de responsabilidad civil de los centros sanitarios donde se vaya a realizar el ensayo incluyan en su cobertura este tipo de ensayos. La mayor parte de los servicios de salud de España ya han lo han incluido, pero es recomendable que durante la fase de preparación del ensayo se confirme esta información con cada uno de los centros sanitarios que se pretendan incluir en la solicitud del ensayo.
2. ¿Implica el estudio a un producto sanitario?
En caso afirmativo, debemos plantearnos 2 opciones: ¿se trata de un estudio observacional o un estudio experimental? La respuesta a esta pregunta se puede responder usando lo indicado con anterioridad para el caso de los medicamentos (pregunta 1).
2.1 Estudios observacionales con productos sanitarios
Es decir, estudios en los que simplemente nos limitamos a observar y recoger datos clínicos relacionados con la utilización de un producto sanitario. Los productos sanitarios tienen marcado CE (Certificado de Conformidad Europeo), se usan en las indicaciones aprobadas y no se modifica la práctica clínica. No existe regulación específica al respecto, pero dado que se trataría de estudios en el ámbito de la biomedicina y siguiendo las recomendaciones generales al respecto como la Declaración de Helsinki3, es recomendable obtener al menos el dictamen favorable de un CEIm acreditado en España así como la conformidad de los centros donde se vaya a llevar a cabo. Como en los estudios observacionales con medicamentos, en esta situación, de nuevo queda a discreción de cada centro el elegir la fórmula de cómo expresar la conformidad y por lo tanto la exigencia o no de la firma de un contrato (especialmente en el caso de que exista una memoria económica asociada).
2.2 Investigaciones clínicas con productos sanitarios
Están definidas en el Real Decreto 1090/20159. Serían aquellos estudios en los que se utiliza un producto sanitario sin marcado CE, o se usa un producto sanitario en indicaciones no aprobadas, o se utilizan procedimientos de diagnóstico o seguimiento a los sujetos de ensayo que van más allá de la práctica clínica habitual.
Es necesario antes del inicio del ensayo la obtención del dictamen favorable de un CEI, la autorización de la AEMPS, y la conformidad de los centros donde se vaya a realizar (de nuevo queda a discreción de cada centro cómo expresar esta conformidad).
En el caso de investigaciones clínicas con productos sanitarios que ostenten el marcado CE y se utilicen en las indicaciones aprobadas en el procedimiento de evaluación de la conformidad, pero en las que se lleve a cabo alguna intervención que modifique la práctica clínica habitual, se notificará su inicio a la AEMPS.
El resto de consideraciones éticas y metodológicas serán las que se aplican para ensayos clínicos con medicamentos. A este respecto cabe mencionar que las condiciones para tener un seguro que cubra los riesgos y perjuicios derivados del estudio serían las mismas que se aplican a los ensayos clínicos. De manera que solo si el producto ostenta marcado CE, se utiliza en las indicaciones contempladas en la conformidad y además todos los procedimientos complementarios de diagnóstico o seguimiento que se llevan a cabo siguiendo el protocolo del ensayo implican un riesgo o carga adicional para la seguridad de los sujetos que es mínimo comparado con el de la práctica clínica habitual en esa población, podría no ser necesario la contratación de un seguro específico. Puede decirse que son las mismas condiciones que se definen en el punto 1.2 para ensayo clínico (con medicamentos) de bajo nivel de intervención. La valoración de este aspecto (necesidad o no de seguro específico) entra dentro de las funciones del CEIm. Aunque, por experiencia, es importante que se deje claro en el protocolo estos puntos para facilitar la evaluación.
3. Nuestro estudio no implica ni medicamentos ni productos sanitarios.
3.1 ¿Recoge muestras biológicas de origen humano o se realizan procedimientos invasivos?
En caso afirmativo, se trata de un estudio al que le aplica lo regulado en la Ley 14/2007, de 3 de julio, de Investigación biomédica12 y el Real Decreto 1716/2011, de 18 de noviembre, por el que se establecen los requisitos básicos de autorización y funcionamiento de los biobancos con fines de investigación biomédica y del tratamiento de las muestras biológicas de origen humano, y se regula el funcionamiento y organización del Registro Nacional de Biobancos para investigación biomédica13. Para este tipo de estudios se ha de obtener el dictamen favorable del CEI del centro donde se llevará a cabo la investigación. En el caso de estudios multicéntricos, para las investigaciones con procedimientos invasivos, en la ley se menciona que si la investigación se realiza en varios centros se garantizará la unidad de criterio y la existencia de un dictamen único. Sin embargo, la práctica nos indica que a día de hoy aún se sigue exigiendo el dictamen del CEI que tutela a cada uno de los centros donde se va a realizar. Lo mismo sería aplicable para estudios que implican la utilización de muestras biológicas.
Además, en estudios que impliquen algún procedimiento invasivo en el ser humano la norma prevé que debe ser autorizada por el órgano autonómico competente. La norma define procedimiento invasivo como toda intervención realizada con fines de investigación que implique un riesgo físico o psíquico para el sujeto de investigación.
Cabe resaltar que, en el caso de estudios con procedimientos invasivos, la norma prevé la contratación de un seguro específico para cubrir los potenciales daños o perjuicios que puedan sufrir los sujetos en investigación. De nuevo la valoración de este aspecto (necesidad o no de seguro específico) entra dentro de las funciones del CEI. Aunque, al igual que se mencionó en el punto previo, por experiencia, es importante que se deje claro en el protocolo el riesgo o carga adicional que pueden implicar el/los procedimientos invasivos para facilitar esta evaluación. Para este análisis, se recomienda la lectura de un artículo publicado en 201514 en el que muy acertadamente se hace un análisis de la consideración de procedimiento invasivo y de cuando este puede suponer o no una carga/riesgo adicional para el participante en el estudio.
En cuanto a la obtención de la conformidad de los centros donde se vayan a realizar, si bien la ley no lo menciona, es recomendable que se consulte a cada uno de los centros por si estos hubieran previsto algún procedimiento local específico en este sentido.
Si las muestras proceden de un biobanco autorizado en España, además del dictamen del CEI del centro donde se realizará la investigación, el biobanco como procedimiento interno solicitará los informes de los 2 comités externos (científico y el ético) que tienen que tener según la normativa que los regula9.
Por último, si en el estudio las muestras a emplear son procedentes de células y tejidos de origen embrionario humano entraría en juego la Comisión de Garantías para la Donación y Utilización de Células y Tejidos Humanos, órgano colegiado adscrito al Instituto de Salud Carlos III. Estos estudios requerirán el informe previo favorable de la citada comisión.
3.2 ¿Nuestro estudio no recoge ni muestras biológicas de origen humano ni se realizan procedimientos invasivos?
Este tipo de estudios no están regulados específicamente en España, pero dado que se trataría de estudios en el ámbito de la biomedicina y siguiendo las recomendaciones generales al respecto como la Declaración de Helsinki, es recomendable obtener al menos el dictamen favorable de un CEI acreditado en España, así como la conformidad de los centros donde se vaya a llevar a cabo. Además, es importante recordar que a la hora de la publicación de los resultados del estudio, la revista exige que los autores declaren o envíen copia del dictamen del comité de ética que revisó el proyecto siguiendo las Recommendations for the Conduct, Reporting, Editing, and Publication of Scholarly Work in Medical Journal15. Los estudios que entran en esta definición son, por ejemplo, estudios observacionales de recogida de información clínica durante el propio proceso de atención y seguimiento de los pacientes en los centros sanitarios (y que no implican medicamentos ni productos sanitarios). En este caso, de nuevo, dependerá de cada centro y de los procedimientos que tenga establecidos, el que se exija que independientemente de que el estudio ya cuente con el dictamen de un CEI, sea obligado a ser presentado o evaluado también por su propio comité.
ConclusionesLa investigación con seres humanos, sus muestras y/o sus datos está, como no podía ser de otra manera, muy regulada. Hasta 4 normas distintas establecen el marco directamente aplicable a estos estudios si bien todas ellas siguen una filosofía similar: dictamen de un comité de ética que evalúe la adecuación del estudio a los estándares científicos, éticos y legales, conformidad (contrato) de los centros donde se vaya a realizar para que estos tengan conocimiento de los estudios que se hacen en su ámbito, y finalmente autorización del organismo competente en aquellos estudios que impliquen, bien mayor riesgo sobre los participantes (ensayos clínicos con medicamentos o productos sanitarios o estudios con procedimientos invasivos), bien mayores repercusiones sobre el sistema de salud (estudios postautorización de seguimiento prospectivo).
FinanciaciónSe declara la no existencia de financiación alguna.
Conflicto de interesesSe declara la no existencia de conflicto de intereses.
