





La esclerosis múltiple es una de las principales enfermedades desmielinizantes del sistema nervioso central. Tiene un importante impacto económico y social. Su etiología no está clara, aunque se barajan varias hipótesis, como la infecciosa o la genética. En su fisiopatología parece que una activación inmunitaria atacaría a las vainas de mielina, causando degeneración axonal progresiva e irreversible.
La enfermedad produce síntomas muy variados, y su diagnóstico exige el cumplimiento de una serie de criterios y la exclusión de otras posibles causas. Además, es muy importante el papel de la neuroimagen, sobre todo la RMN.
A pesar de disponer de fármacos modificadores de la enfermedad, todavía no hay ninguno que frene su evolución, y los fármacos útiles son los orientados a paliar la sintomatología de los brotes.
En conjunto, la esclerosis múltiple requiere un importante esfuerzo de investigación que permita aclarar no solo por qué y cómo se produce, sino también el desarrollo de nuevas medidas que mejoren la vida de los pacientes afectados.
Multiple sclerosis is a major demyelinating disease of the central nervous system. It has a significant economic and social impact. Its etiology is unclear, although there are several hypotheses, such as infections or genetics. In its pathophysiology, it seems that immune activation attacks the myelin sheath, causing a progressive and irreversible axonal degeneration.
The disease produces a variety of symptoms, and diagnosis requires fulfilling a number of criteria and the exclusion of other possible causes. The role of neuroimaging, especially MRI, is very important.
Despite the availability of disease-modifying drugs, none of them are able to halt its progress, and the most useful drugs are those designed to alleviate the symptoms of outbreaks.
Overall, multiple sclerosis requires a significant effort in research to clarify not only why and how it occurs, but also to develop of new measures to improve the life of affected patients.
La esclerosis múltiple (EM) es el principal trastorno del grupo conocido como enfermedades desmielinizantes del sistema nervioso central. Actualmente es la primera causa de discapacidad neurológica en adultos jóvenes en los países desarrollados, y su incidencia va en aumento. Esto comporta un alto coste, no solo económico, sino también humano.
A esto se le suma el hecho de que, aunque su patogenia está establecida en parte, los mecanismos que la producen no se conocen del todo bien, y esto imposibilita el establecimiento de un tratamiento eficaz.
Por tanto, comprender la etiología y desarrollar un adecuado diagnóstico y tratamiento son objetivos muy importantes a conseguir para reducir el impacto que puede llegar a tener esta enfermedad en un futuro.
Caso clínicoPaciente mujer de 33 años que acude a su médico de cabecera por presentar cuadro de 2 meses de evolución de parestesias y pérdida de fuerza en la pierna derecha que le ocasiona problemas en la marcha. Describe sensación de que cuando camina y desciende escaleras no nota el suelo. Además, refiere visión borrosa desde hace una semana, junto con fatiga intensa que se hace más pronunciada al caer la tarde. No presenta otros síntomas y niega tener alguna enfermedad o antecedentes de interés.
Exploración física: en la exploración de los pares craneales se detecta un defecto pupilar aferente y una ligera desviación del ojo izquierdo, que presenta dificultad para la aducción. El resto de los pares son normales. La exploración de la extremidad inferior derecha muestra una pérdida de fuerza grado 3/5. Existe un pequeño aumento del tono muscular e hiperreflexia. No se encuentran alteraciones en las pruebas de coordinación y equilibrio.
Pruebas complementarias: se solicita estudio TC junto con potenciales evocados de vías aferentes y eferentes. En la TC no se encuentran hallazgos de interés. Los potenciales evocados muestran afectación de la vía piramidal y visual. Además, se observa una afectación asintomática de la vía auditiva.
Dados los resultados de las pruebas diagnósticas se deriva a la paciente a Neurología, donde se le realiza una resonancia magnética con gadolinio. En esta se observan múltiples áreas hiperintensas en la sustancia blanca subcortical, así como afectación de varios segmentos de la médula espinal, compatibles con zonas de inflamación. Además, se extrae una muestra de LCR para análisis. Los resultados revelan una ligera pleocitosis, junto con presencia de bandas oligoclonales.
Diagnóstico: sin otras causas que expliquen esta afectación, se diagnostica a la paciente de EM probable.
EtiologíaAunque no hay nada establecido al respecto, las líneas de investigación abiertas para entender el origen de esta enfermedad son múltiples. Lo que es cierto es que su etiología es multifactorial, dependiente de la interacción entre genes y factores ambientales.
La carga genética de la enfermedad es indiscutible, y se ha demostrado la relación con el HLA de clase ii, siendo DRB1*15:01 el alelo con una susceptibilidad más elevada. No obstante, también se pueden encontrar alelos protectores, sobre todo en el HLA de clase i. La interacción entre los genes protectores y los de susceptibilidad es lo que favorecerá el desarrollo de la EM, su expresión fenotípica y su evolución a lo largo del tiempo1.
En cuanto a los factores de riesgo, se postula el efecto de la vitamina D sobre la enfermedad. Parece que niveles bajos están relacionados con el desarrollo de EM, así como con un peor pronóstico de la enfermedad, pero no son datos concluyentes2.
El componente infeccioso siempre se ha tenido en cuenta, pero no se ha encontrado el agente causante. El virus de Epstein-Barr es uno de los candidatos, puesto que se han hallado altos niveles de anticuerpos contra este virus en un alto porcentaje de individuos con EM3. Este riesgo es más elevado cuanto mayor sea la persona al contraer la infección, así como tener clínica mononucleósica también implica ligeramente más riesgo de EM que aquellas formas asintomáticas4. Sin embargo, no solo este virus puede ser uno de los causantes, ya que también se hallan elevados anticuerpos contra otros microorganismos, como sarampión, varicela zóster o virus herpes simple.
Finalmente, el tabaco es el otro gran factor de riesgo que se estudia. Tampoco hay resultados que lo acaben de confirmar, pero parece que favorece el paso a formas clínicamente definidas de EM. A esto se le suma el hecho de que los pacientes fumadores tienen una importante atrofia cerebral, junto con una inflamación más activa, lo que conduce a un mayor deterioro a largo plazo5.
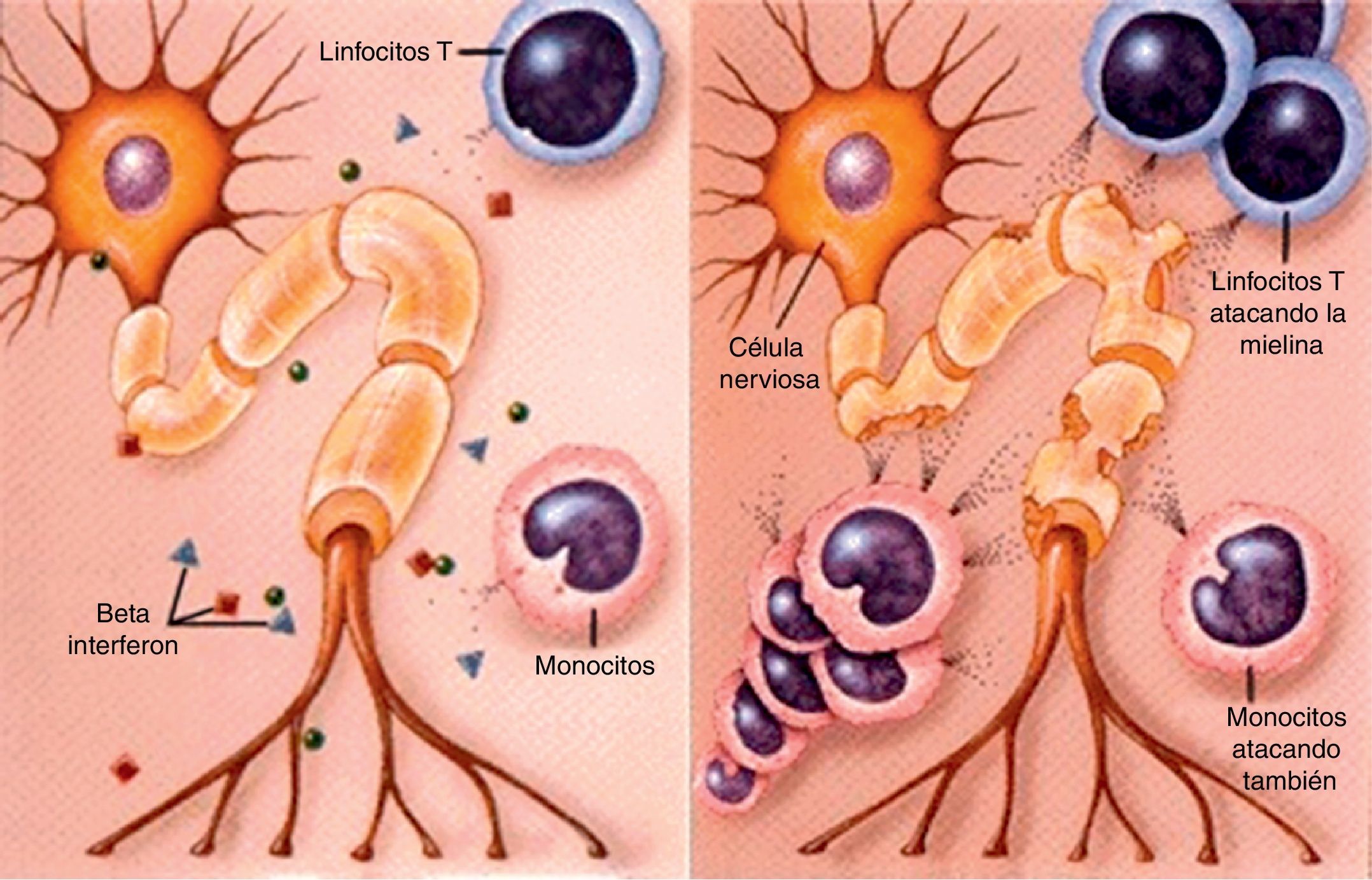
Fisiopatología6,7La EM es una enfermedad desmielinizante del sistema nervioso central provocada por la destrucción de las vainas de mielina por parte del sistema inmunitario. Esto es debido a la existencia de un infiltrado inflamatorio formado por macrófagos, linfocitos T CD8 y microglia que ataca a los oligodendrocitos, ya sea de forma directa o a través de sustancias como radicales de oxígeno o glutamato. También los linfocitos B proliferan, dando lugar a una expansión clonal con producción de anticuerpos antimielina.
Cuando las lesiones son importantes se activa la proliferación de los astrocitos, que conducirá a la gliosis del tejido nervioso (fig. 1). De este modo, la EM se puede resumir en inflamación-desmielinización-gliosis.
 y el de un enfermo de E.M.")
En cada episodio inflamatorio se produce pérdida axonal, aunque no está claro si la destrucción de mielina es un paso necesario para provocarla. En un inicio los axones pueden adaptarse promoviendo la remielinización, pero, eventualmente, se produce degeneración distal y retrógrada. El acúmulo de axones destruidos es lo que lleva a las manifestaciones clínicas de la enfermedad.
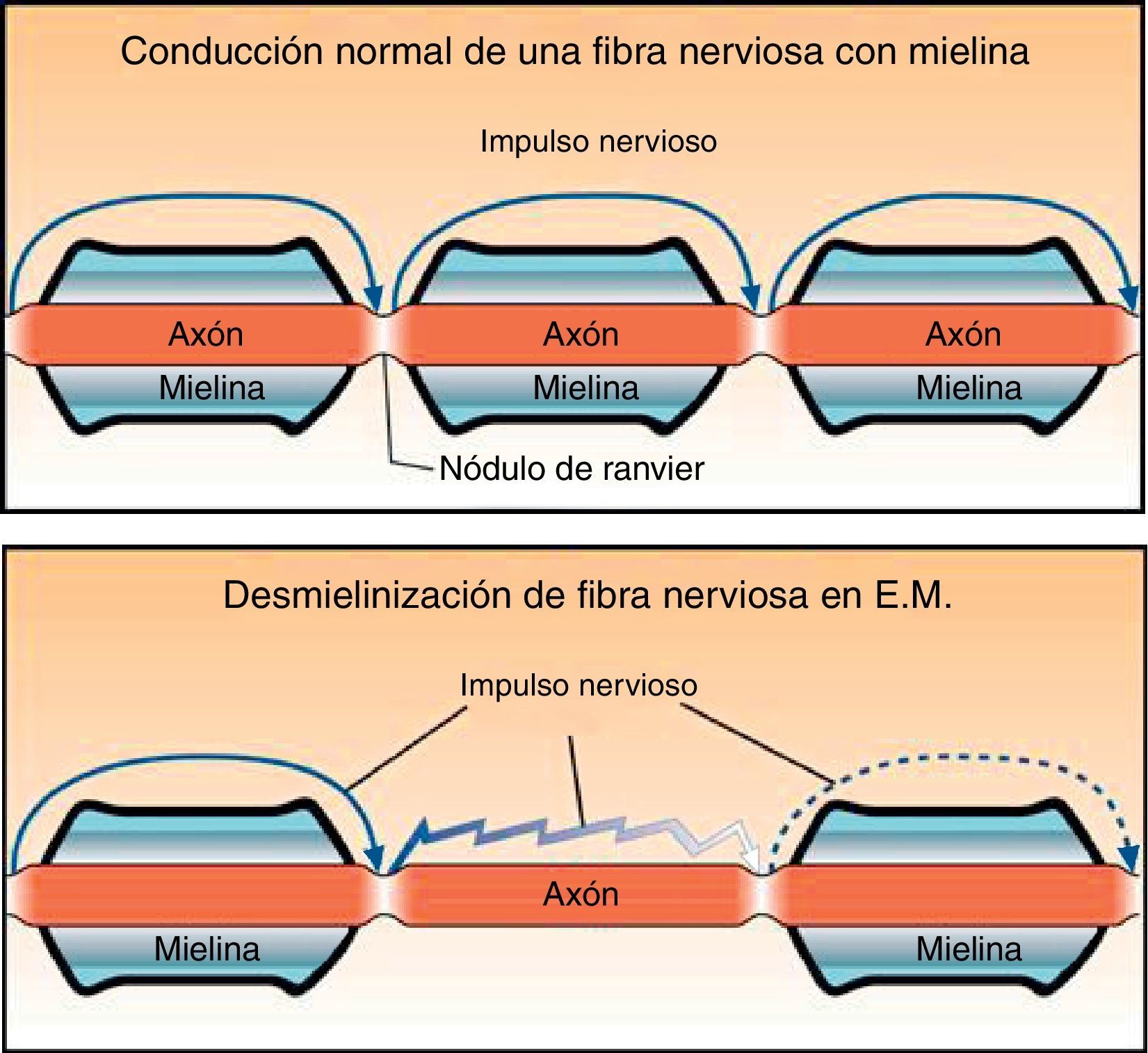
Sin la protección de mielina la membrana axonal queda descubierta, lo que genera cambios en el potencial de acción de membrana y en la distribución de los canales iónicos (fig. 2). En condiciones normales el impulso nervioso se transmite de forma saltatoria de un nódulo de Ranvier a otro, lo que permite velocidades de conducción muy elevadas. Sin mielina la conducción es continua y más lenta.
 y uno con pérdida de mielina en la esclerosis múltiple (abajo).")
Además, pueden ocurrir bloqueos de la conducción, sobre todo en situaciones de hipertermia (fiebre o ejercicio) y en determinadas alteraciones metabólicas. También es frecuente al inicio de la desmielinización, cuando los canales de sodio todavía no se han extendido a las zonas descubiertas.
Todas estas alteraciones se producen en diferentes regiones del sistema nervioso central a lo largo de varios episodios o de forma continua, dependiendo del tipo de evolución de la enfermedad.
EpidemiologíaLa prevalencia de EM es mucho más elevada en países desarrollados y afecta mucho más a las mujeres. De hecho, en los últimos años, la prevalencia está aumentando sobre todo en este grupo de población. Generalmente se inicia entre los 20-40 años de edad, pero se pueden encontrar casos en las edades extremas de la vida.
Tiene una distribución geográfica particular, ya que aumenta en las latitudes más altas. De este modo, Escandinavia, Canadá, EE. UU. o Nueva Zelanda son zonas de elevada prevalencia. No obstante, nuevos estudios parecen mostrar que este efecto de la latitud está desapareciendo8.
La relación con los movimientos migratorios también es importante, puesto que parece que los individuos que migran de una zona de bajo riesgo a una de alto incrementan la probabilidad de desarrollar la enfermedad.
ClínicaLas manifestaciones de la EM son muy variables, ya que dependen de la zona del sistema nervioso donde se producen las lesiones. Se pueden iniciar tanto de forma brusca como ir evolucionando progresivamente9,10.
La debilidad muscular es uno de los síntomas más frecuentes, y suele iniciarse en una de las extremidades inferiores, aunque también puede darse en forma de debilidad facial. Generalmente se acompaña de afectación piramidal con hiperreflexia, signo de Babinski y espasticidad. Puede haber pérdida de reflejos tendinosos si se afecta la médula espinal. En cuanto a la espasticidad, aparece de forma espontánea o con el ejercicio, y puede ser muy dolorosa. Sumada a la debilidad muscular, la alteración de la marcha es importante.
No solo hay afectación motora. La alteración sensitiva en forma de parestesias o hipoestesia también es frecuente, incluso más que la alteración motora. En otras ocasiones los pacientes notan sensaciones desagradables, y un gran número presenta episodios de dolor que pueden afectar a cualquier parte del cuerpo.
La mayoría de los pacientes experimenta fatiga, siendo la principal causa de incapacidad laboral. Puede sentirse como un cansancio constante y verse agravado por el ejercicio físico. Al caer la tarde suele empeorar también.
Los episodios de neuritis óptica provocan una disminución de la agudeza visual que, si bien no suele llegar a la ceguera total, puede alterar gravemente la visión. Habitualmente afecta a un solo ojo y se acompaña de dolor orbitario que se incrementa con los movimientos oculares. Puede haber un defecto pupilar aferente del ojo afecto, y en la exploración, el fondo de ojo puede ser tanto normal como observarse una papilitis.
Otra afectación ocular frecuente es la oftalmoplejía internuclear, que produce dificultad de aducción del ojo afecto junto con nistagmo. El hallazgo en ambos ojos es altamente indicativo de EM11.
La afectación cerebelosa conduce a ataxia y temblores tanto de la cabeza como del cuerpo, e incluso a una disartria característica. La disfunción vesical es otro síntoma frecuente (tabla 1).
Criterios diagnósticos de McDonald (2001)
| Presentación clínica | Datos adicionales necesarios |
|---|---|
| 2 o más brotes; 2 o más lesiones clínicas objetivas | Ninguno, la evidencia clínica es suficiente |
| 2 o más brotes; una lesión clínica objetiva | Diseminación en espacio demostrada por: RMN o LCR positivo y 2 o más lesiones en la RMN compatibles con EM o nuevo ataque clínico que afecte un sitio diferente |
| Un brote; 2 o más lesiones clínicas objetivas | Diseminación en tiempo demostrada por: RMN o segundo ataque clínico |
| Un brote; una lesión clínica objetiva | Diseminación en espacio demostrada por: RMN o LCR positivo y 2 o más lesiones en la RMN compatibles con EM y diseminación en tiempo demostrada por: RMN o segundo ataque clínico |
| Clínica indicativa de EM progresiva primaria | LCR positivo y diseminación en espacio demostrada por (al menos uno): evidencia en RMN de 9 o más lesiones cerebrales en T2; 2 o más lesiones en médula espinal; 4-8 lesiones cerebrales y una lesión de médula espinal; PE positivos con 4-8 lesiones en la RMN; PE positivos con menos de 4 lesiones cerebrales y más de una lesión en la médula espinal y diseminación en tiempo demostrada por: RMN o progresión continuada por un año |
El deterioro cognitivo no suele ser una forma de inicio de la EM, pero sí es un síntoma muy prevalente (hasta el 50% de los casos). Es un deterioro que, a diferencia del que se da en la enfermedad de Alzheimer, sigue un patrón de tipo subcortical, con afectación de algunas áreas como atención, fluencia verbal, memoria, razonamiento abstracto, percepción visuoespacial, resolución de problemas y formación de conceptos, y velocidad de procesamiento de datos. La afectación de la memoria no correlaciona con la progresión de la EM, con la discapacidad física, con el tiempo de evolución, ni tampoco con la depresión. No se ha encontrado correlación con factores demográficos, pero sí se ha hallado entre la severidad de la afección cerebral estudiada por RM y el deterioro cognitivo12. Por ello, las pruebas de cribado que se utilizan habitualmente en Atención Primaria, como el Mini-Examen Cognoscitivo o el MMSE no son útiles, y una correcta valoración del deterioro cognitivo precisaría baterías neuropsicológicas amplias. Además, otros síntomas que aparecen en la EM, como la depresión o la fatiga, pueden afectar al rendimiento cognitivo y a la manera en que este se mida13.
Dada la amplia variedad de lesiones en el sistema nervioso, la lista de síntomas se alarga mucho más, aunque la proporción de pacientes que los presentan es muy pequeña: signo de Lhermitte, neuralgias, disfunción y sexual, fenómeno de Uhthoff.
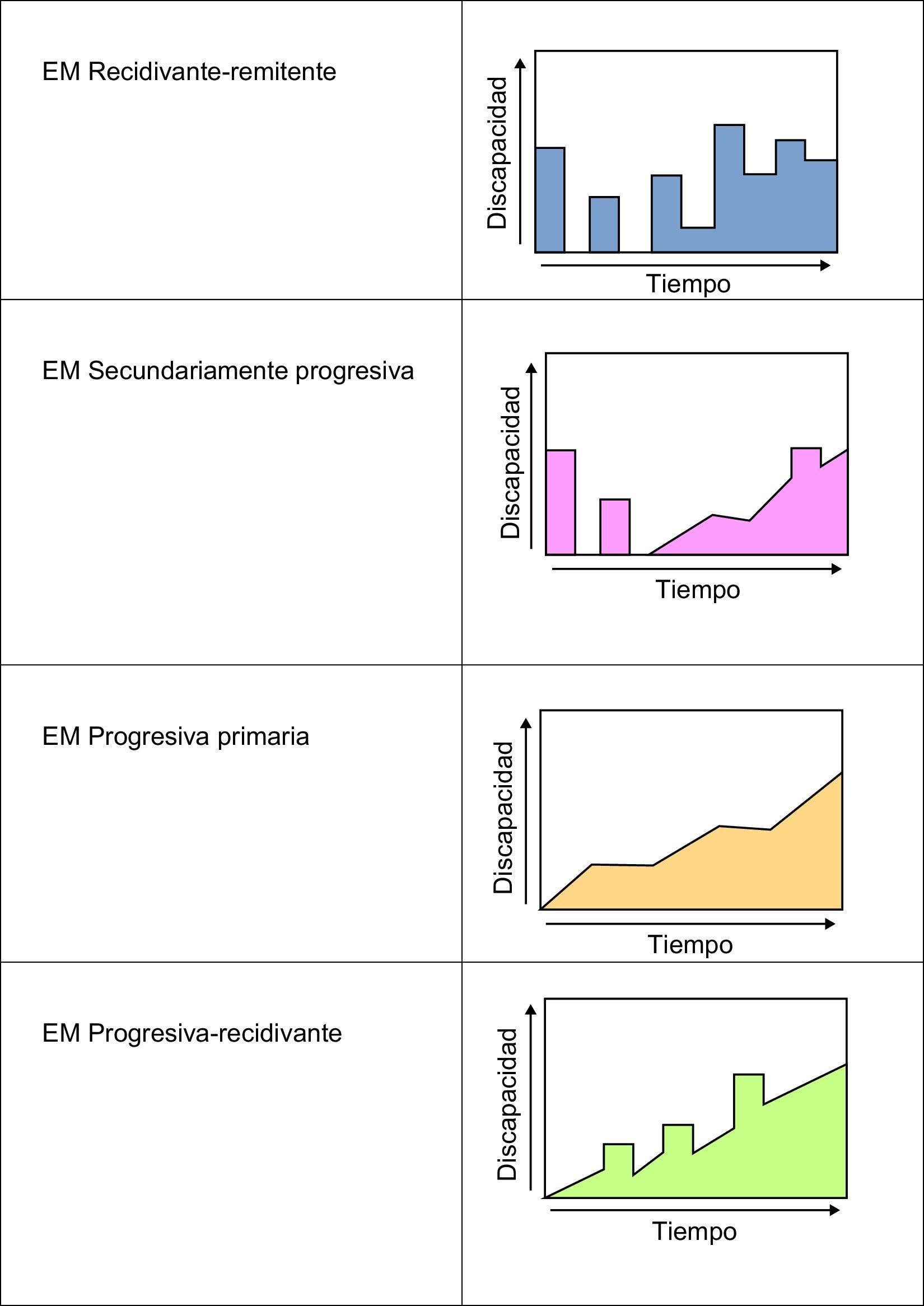
Variantes en la evolución de la esclerosis múltipleLa EM presenta diferentes pautas de progresión. Esto permite dividirla en varios tipos, que se enumeran a continuación (fig. 3):

Recidivante-remitente (85%): produce crisis que se alargan varios días e incluso semanas, con periodos estables intercríticos.
Secundariamente progresiva: se trata de la evolución de muchos pacientes con EM recidivante-remitente en la que, con el paso del tiempo, se va instaurando afectación mielínica permanente y deterioro continuo. En ocasiones los pacientes pueden volver a presentar brotes.
Progresiva primaria (15%): la evolución se produce de forma progresiva y constante, sin crisis de empeoramiento. La incapacidad acontece de forma más temprana. Esta variante es proporcionalmente más frecuente en hombres, y suele iniciarse alrededor de los 40 años.
Progresiva-recidivante: esta variante se inicia como una progresiva primaria que, en cambio, registra brotes superpuestos al deterioro continuo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
