


Reduced sperm motility represents one of the major male causes of infertility. Ultrastructural defects in the sperm flagellum caused by genetically inherited and congenital defects are one of the main causes to reduced sperm immotility. Several molecular components have been already associated to reduced sperm motility and more are expected to be discovered, especially with the application of Next-Generation Sequencing technology. In this review we will give emphasis to the main molecular components of the sperm flagellum associated to sperm motility. We will also discuss some of ultrastructural defects in structures of sperm flagellum and the two main genetic disorders that are associated with poor sperm motility: Primary Ciliary Dyskinesia and Dysplasia of the Fibrous Sheath, with reference to genes that are known to be involved in these disorders.
La reducción de la movilidad espermática constituye una de las principales causas de infertilidad masculina. Los defectos ultraestructurales en el flagelo, derivados de defectos genéticos y congénitos, son una de las principales causas de la inmovilidad espermática. Son varios los componentes moleculares asociados a una menor movilidad espermática y es de esperar que se descubran otros con la aplicación de nuevas técnicas de secuenciación. En esta revisión nos centraremos en los principales componentes moleculares del flagelo asociados a la movilidad. También analizamos algunos de los defectos ultraestructurales en la estructura del flagelo y los dos principales trastornos genéticos que se asocian a la movilidad espermática deficiente: la discinesia ciliar primaria y la displasia de la vaina fibrosa, con referencia a los genes involucrados en dichos trastornos.
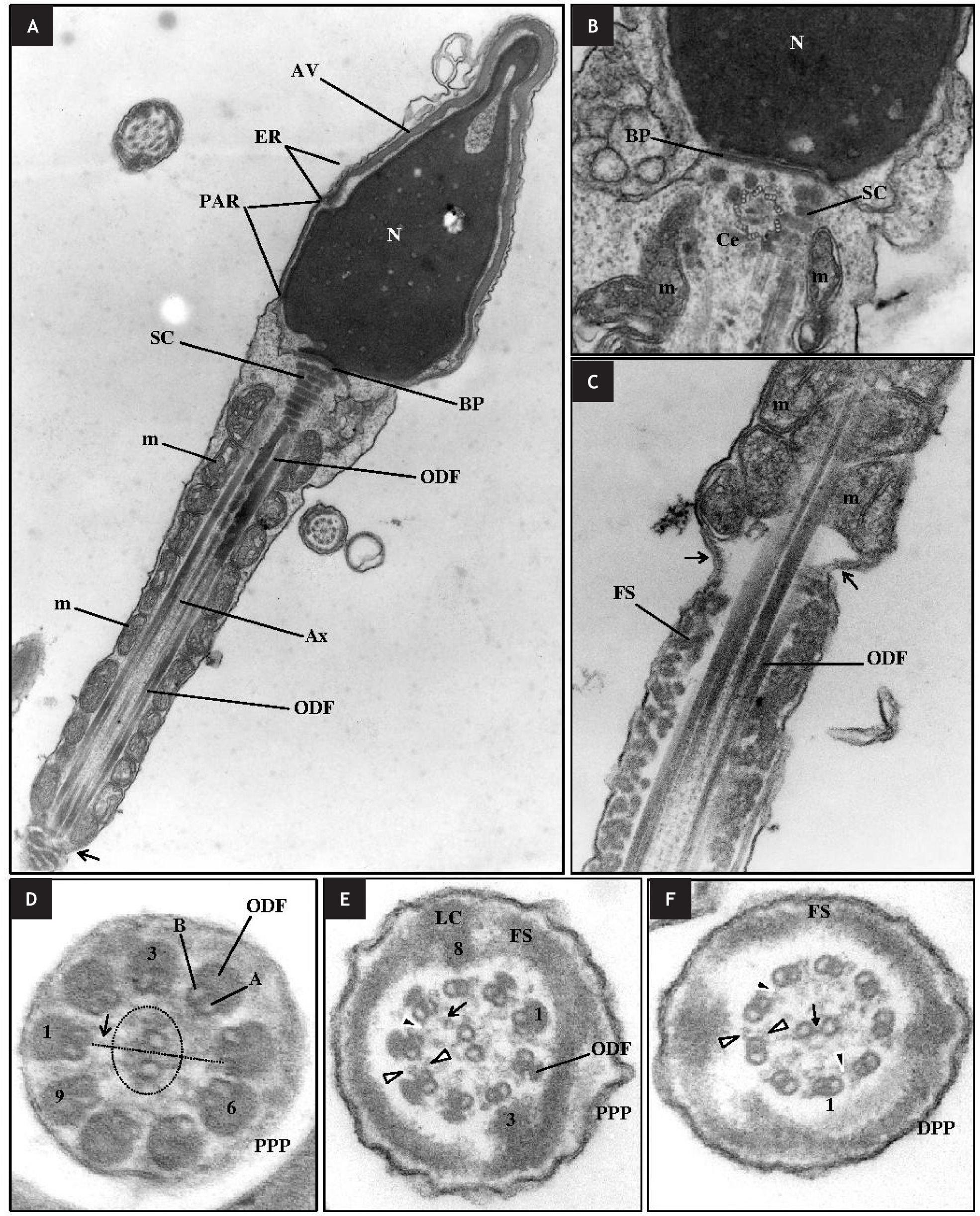
The spermatozoon (Figs. 1A-1C) is divided into two fundamental parts, the sperm head and the sperm tail or flagellum. The main components of the sperm head are the nucleus, which contains the genetically material, and the acrosomal vesicle, which covers the anterior half of the head and contains crucial enzymes for the acrosomal reaction and is of great importance for fertilization. The flagellum is responsible for sperm motility and contains both the energy production site and the propulsive apparatus of the cell. The flagellum consists of four distinct segments: the neck piece (NP), the midpiece (MP), the principal piece (PP) and the end piece (EP). The NP contains the basal plate (BP), the proximal centriole (PC) and the striated/segmented columns (SC). The MP contains the axoneme (Ax), the outer dense fibers (ODF) and the mitochondria sheath. The PP contains the Ax, the ODF (proximal PP) and the fibrous sheath (FS: proximal and distal PP). The PP is separated from the MP by the annulus (An) that is a ring of dense material found at the end of the mitochondrial sheath. The EP contains only the Ax.1,2
; acrosomal vesicle (AV) with its final region, the equatorial region (ER); posterior acrosomal region (PAR) that is the region of the head without the AV; neck region with the basal plate (BP), centriole (Ce), axoneme (Ax) and striated columns (SR); midpiece with outer dense fibers (ODF) and the mitochondrial sheath (m). This region ends at the annulus (arrow). C. The annulus (arrows) separates the midpiece from the principal piece where the fibrous sheath (FS) begins. D. The axoneme at the proximal principal piece (PPP) is adjacent to 9 ODF. Each doublet is formed by the A and B microtubules. Doublets are numbered in a clockwise direction. The central pair is surrounded by a fibrilar sheath (dotted circle). Each doublet has a radial spoke (arrow). E. Outer and inner dynein arms (white arrowheads); nexin bridges (black arrowhead). F. Distal principal piece (DPP). The central pair is connected by a central bridge (arrow).")
Ultrastructure of the normal spermatozoon. A, B. Nucleus (N); acrosomal vesicle (AV) with its final region, the equatorial region (ER); posterior acrosomal region (PAR) that is the region of the head without the AV; neck region with the basal plate (BP), centriole (Ce), axoneme (Ax) and striated columns (SR); midpiece with outer dense fibers (ODF) and the mitochondrial sheath (m). This region ends at the annulus (arrow). C. The annulus (arrows) separates the midpiece from the principal piece where the fibrous sheath (FS) begins. D. The axoneme at the proximal principal piece (PPP) is adjacent to 9 ODF. Each doublet is formed by the A and B microtubules. Doublets are numbered in a clockwise direction. The central pair is surrounded by a fibrilar sheath (dotted circle). Each doublet has a radial spoke (arrow). E. Outer and inner dynein arms (white arrowheads); nexin bridges (black arrowhead). F. Distal principal piece (DPP). The central pair is connected by a central bridge (arrow).
The Ax (Figs. 1D-1F) is the flagellar motor. Its basic structure is represented by a 9d+2s microtubule pattern, with a pair of central microtubules (MT), C1 and C2, which are surrounded by nine peripheral MT doublets. The Ax is surrounded by the ODF and then by mitochondria in the MP, by the ODF and then by the FS in the proximal PP, whereas in the distal PP it is only surrounded by the FS.2
The nine peripheral doublets are numbered 1 to 9 in a clockwise direction (number one is the one perpendicular to the central pair of MT). Each doublet consists of an internal complete MT, A, onto which is attached a second external and incomplete MT, B. Microtubule A has two dynein arms, outer (ODA) and inner (IDA). Doublets are linked to each other by nexin bridges and to the central pair of MT by the radial spokes. Nexin bridges act as a regulator of the dynein complex and structurally limits doublet sliding.3 The two MT of the central pair are linked by a series of regularly spaced linkages (central bridge) and are surrounded by a fibrilar central sheath that are formed by a pair of spiral fibres attached to the central MT at the level of the connecting links. These constitute the central apparatus of the Ax.1,4 Each MT doublet is externally anchored to 9 corresponding asymmetric ODF1,4 that protect the tail against shearing forces encountered during epididymis transport and especially during ejaculation, but also during transit through the female genital tract.5
The molecular composition of flagellum components has been studied mainly in sperm from marine invertebrates and the biflagellate green algae Chlamydomonas. These showed that the molecular composition of the flagellum components is composed of approximately 250 proteins. The Ax is a sophisticated structure with a cytoskeleton, protein motors, molecular chaperones, regulatory elements such as Ca2+ binding proteins and protein kinases/phosphatases.6,7.
Tubulins α and β are the main constituents of MT. These globular proteins of 50–55 kDa constitute 70% of the protein mass of the Ax.8,9 Tubulins are often subjected to post-translational modifications, such as acetylation, palmitoylation, phosphorylation, polyglutamylation and polyglycation,10 which are important for proper binding and assembly of the axoneme MT and motility.11 For instance, polyglutamylation of α-tubulin plays a dynamic role in the dynein-based motility process.12
Another essential class of Ax proteins are dyneins. Dyneins are ATPases from a family of motor proteins that drive microtubule sliding in cilia and flagella.13 These motor proteins convert the chemical energy contained in ATP into the mechanical energy of movement. Dyneins can be divided into two groups: cytoplasmic dyneins and axonemal dyneins. The axonemal dyneins are key elements to motility of eukaryotic cilia and flagella and comprise the ODA and the IDA. The ODA is composed of two heavy chains (HC), α and β; three to five intermediate chains (IC) and six light chains6 (LC). It produces most of the force for flagellar movement.14 The IDA are more complex, with eight distinct HC, which are organized with various IC and LC into seven different molecular complexes, one two-headed isoform and six single-headed isoforms.15 The HC contain the motor machinery that is responsible for transducing chemical energy into directed mechanical force applied to the microtubule surface, possessing the sites of both ATP hydrolysis and ATP-sensitive microtubule binding.16,17 The IC and LC are thought to be involved in binding dynein to MT-A.16 They also help to specify the intracellular location of the dynein and regulate its motor activity.17,18 In response to changes in motility they are also regulated through phosphorylation/dephosphorylation through a kinase/phosphatase system present in the radial spoke and central pair.6
The ODA Docking Complex (ODA-DC) is a structure that interacts directly with the ODA and is responsible for its assembly at regular intervals of 24 nm. It is also important as an intermediate in the binding of ODA to its unique attachment site within MT-A.14 The ODA-DC contains three polypeptides (DC1-DC3). The DC1 and DC2 polypeptides potentially determines the 24-nm longitudinal spacing of the ODA.14 The DC3 polypeptide has some important roles in the regulation of the ODA, playing a role in calcium-regulated ODA activity.19
The Dynein Regulatory Complex (DRC) is composed of six Ax proteins.20 Studies using DRC mutants showed that some components of the DRC serve primarily to regulate activity, while others play a role in mediating structural interactions between dynein arms, the A-tubule of the outer doublet, and the radial spokes.13,20 Recent studies using cryo-electron tomography, revealed that DRC forms a continuous connection from the A-tubule to the B-tubule of the neighbouring microtubule doublet.3 This continuous connection and the finding that the DRC is the only structure besides the dynein arms that connects with adjacent outer doublets led the authors to suggest that the DRC is the nexin link and to propose the term nexin-DRC (N-DRC) to the DRC.3
The radial spokes and central pair are essential structures for the regulation of dynein arms.21,22 Among other important roles, it was proposed that radial spokes and central apparatus may be involved in converting simple symmetric bends into the asymmetric waveforms required for forward swimming and in the release of ATP inhibition in a controlled manner.21 In Humans it has been already described at least seven radial spoke proteins that have several isoforms: RSPH4A, RSPH6A, RSPH3, RSPH9, RSPH10B2, RSPH10B and RSPH1.23 The central pair functions like a distributor to provide a local signal to the radial spokes that selectively activates subsets of dynein arms.24
The Fibrous SheathThe Fibrous Sheath (Fig. 1) is a unique characteristic of the spermatozoon and consists of two peripheral longitudinal columns, which are at the plane of the central MT, connected together by a series of ribs. The ribs are composed of closely packed filaments and form a ring around the axoneme.1,2 The FS is believed to influence the degree of flexibility, plane of flagellar motion and the shape of the flagellar beat.25
Three important FS proteins belong to the “cAMP-dependent protein kinase anchoring protein” (AKAP) family.25 AKAP are scaffolding molecules that organize molecular complexes whose function is to modulate signalling pathways. Besides the AKAP family, the FS is composed by other proteins, such as Ropporin, Rhophilin and the “Calcium-binding tyrosine phosphorylation regulated protein” (CABYR),25 which are extremely important for FS assembly and function, and thus for sperm motility. Another important class of proteins that are present in the FS are the glycolytic enzymes. Studies suggest that the delivery of ATP from the mitochondria is not enough to sustain sperm motility and that sperm had to develop alternative methods of energy production that are independent of the mitochondrial oxidative phosphorylation.26,27 The ATP generated from mitochondria is mainly used for membrane changes occurring during maturation in the epididymis, during capacitation in the female genital tract, and for the acrosome reaction. Flagellar glycolysis from the FS is the main producer of the ATP required for axoneme beating. Some of the glycolytic enzymes, including spermatogenic cell-specific forms of two glycolytic enzymes [glyceraldehyde 3-phosphate dehydrogenase (GAPD) and hexokinase 1 (HK1)], are tightly associated with the FS.25-27
Sperm motilityThe Ax is the fundamental structure responsible for motility. Flagellum motility is a consequence from undulatory waves propagating backwards that create forward propulsive thrust along the axis of the flagellum. The flagellar motility, from which the sperm motility arises, is created by the motor activities of the axoneme dynein arms working against the stable microtubule doublets.
Dynein cAMP-dependent phosphorylation leads to an interaction between dynein arms and the microtubule doublet, starting the flagellar beat, activates ATPase activity and begins the conversion of the chemical energy from ATP hydrolysis into mechanical energy for motility. The process is reversed by dephosphorylation of dynein by the calmodulin-dependent protein phosphatase calcineurin.26,28 Therefore, phosphorylation/dephosphorylation have to occur in an asynchronous way through the entire Ax. As AKAP proteins sequester enzymes such as protein kinases and phosphatases with appropriate substrates to the coordination of phosphorylation and dephosphorylation events,29 they could be also involved in those phosphorylation events. Sperm motility is thus a highly complex process with several structural and molecular elements, and metabolic pathways involved.26,28
Flagellar abnormalities and some genetic bases of sperm immotility in humansDue to the highly complexity of sperm motility, any alteration in external and/or internal factors regulating sperm motion, as well as in cellular structure and metabolism involved in generating flagellar beat, may result in defects in sperm motility, which consequently results in male infertility.
Asthenozoospermia (ATZ) is the medical term for reduced sperm motility and is one of the main male pathologies underlying infertility.30,31 The etiology of ATZ is not simple to unravel and often remains unexplained. Ultrastructural defects in the sperm flagellum caused by genetically inherited and congenital defects,32-34 and necrozoospermia (absence of live spermatozoa in the ejaculate), are main causes of ATZ.30,31 Although in the mouse, human mitochondrial sheath and mutations in human mtDNA are other causes of ATZ, mainly due to disruption of the MP.35-37
Besides structuraly separating the MP from the PP, the An maintains sperm membrane domains, and its absence produces an interruption in the cytoskeleton at the MP-PP junction, with disorganization and associated ATZ.38,39 Septins (SEPT) are essential structural components of the An and defects in SEPT are also associated to ATZ.38,40,41
Although in the mouse, mutations in genes coding for several transport, structural, motor and signalling proteins, as well as for transcription factors of the sperm flagellum are known to cause motility disorders,42,43 in humans a strict association between gene mutations and alterations in sperm motility is still very scarce. However, due to the high degree of conservation of many of these genes among mice and humans, some genes, isolated or associated to syndromes, have already been proved to be responsible or are suspected of being responsible for some cases of human infertility associated with poor sperm motility.11 The two main genetic disorders that are associated with poor sperm motility are Primary Ciliary Dyskinesia (PCD) and Dysplasia of the Fibrous Sheath (DFS).
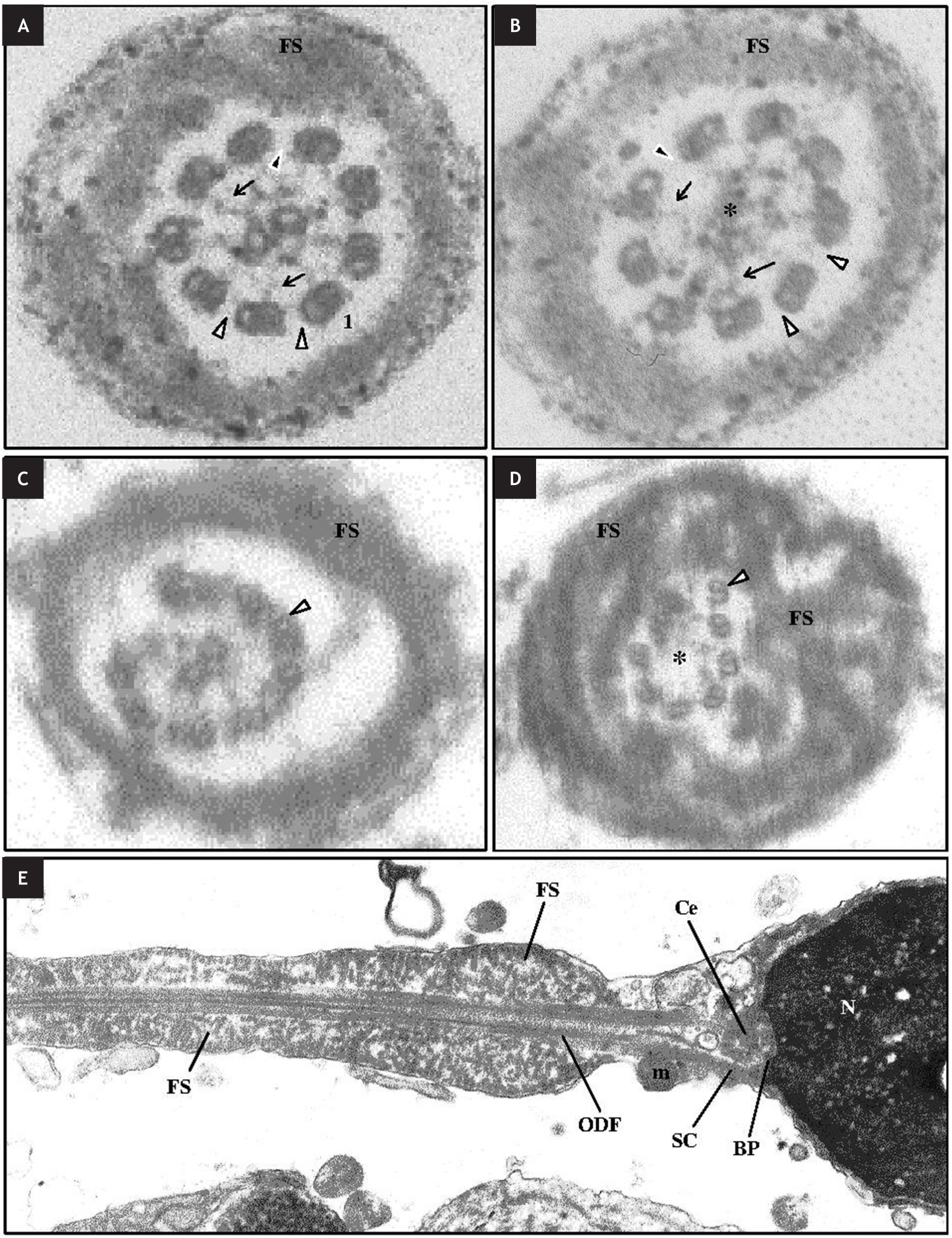
Primary ciliary dyskinesiaPrimary ciliary dyskinesia (PCD, OMIM: 244400), was first described by Afzelius and collaborators.44 Primary ciliary dyskinesia is a genetically heterogeneous, autosomal recessive disease that is characterized by a generalized paralysis of ciliated cells, including sperm and respiratory cilia, resulting in recurrent infections of the respiratory tract. In about 50% of affected individuals situs inversus (a congenital condition in which the major visceral organs are reversed) is present and is known as Kartagener syndrome (KS).45 Most men with PCD have nearly 100% immotile spermatozoa and are consequently infertile. The estimated incidence of PCD is approximately 1 per 15,000 births.46 In the majority of cases, the results of electron microscopic analysis of sperm reveal that the MT doublets lack dynein arms. In some PCD patients were as well detected absence or dislocation of the central MT, defects of radial spokes and peripheral MT abnormalities33,46,47,(Figs. 2A-2C). Besides the ultrastructural defects in sperm cells that leads to sperm immotility, a study also detected a high level of sperm DNA damage in a patient with KS syndrome, which highly reduces the probability of a healthy offspring, even with the application of assisted reproduction techniques.48
 and fibrous sheath (FS) dysplasia (D). A, B. Absence of nexin bridges (black arrowheads) and of the outer and inner dynein arms (white arrowheads), and partial absence of radial spokes (arrows), with presence (A) or absence (*) (B) of the central pair. C, D. Disorganization (C) and displacement (D) of the doublets (white arrowheads), with presence (C) or absence (*) (D) of the central pair. E. Ultrastructure of a spermatozoon with fibrous sheath dysplasia. Note the absence of the annulus and midpiece, with ascension of the dysplastic fibrous sheath (FS). Nucleus (N); basal plate (BP); centriole (Ce); striated columns (SC); mitochondria (m); outer dense fibers (ODF).")
D. Ultrastructure of abnormal axonemes at distal principal piece in sperm of a patient with primary ciliary dyskinesia and situs inversus (A-C) and fibrous sheath (FS) dysplasia (D). A, B. Absence of nexin bridges (black arrowheads) and of the outer and inner dynein arms (white arrowheads), and partial absence of radial spokes (arrows), with presence (A) or absence (*) (B) of the central pair. C, D. Disorganization (C) and displacement (D) of the doublets (white arrowheads), with presence (C) or absence (*) (D) of the central pair. E. Ultrastructure of a spermatozoon with fibrous sheath dysplasia. Note the absence of the annulus and midpiece, with ascension of the dysplastic fibrous sheath (FS). Nucleus (N); basal plate (BP); centriole (Ce); striated columns (SC); mitochondria (m); outer dense fibers (ODF).
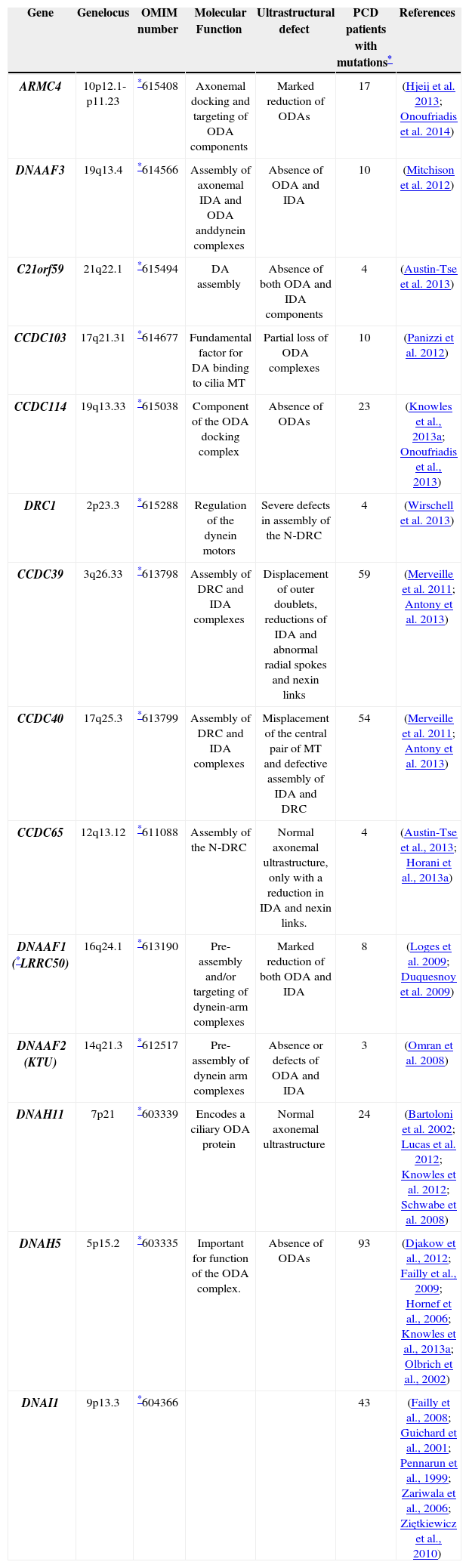
Given that the typical diagnostic of PCD is the absence of dynein arms, the investigations into the genetic basis of PCD have been focused on dynein arm proteins and several genes (Table 1) are known to be associated with PCD.49,50
List of genes known to be associated with Primary Ciliary Dyskinesia (PCD) and main ultrastructural defects found in the axoneme of PCD patients.
| Gene | Genelocus | OMIM number | Molecular Function | Ultrastructural defect | PCD patients with mutations* | References |
|---|---|---|---|---|---|---|
| ARMC4 | 10p12.1-p11.23 | *615408 | Axonemal docking and targeting of ODA components | Marked reduction of ODAs | 17 | (Hjeij et al. 2013; Onoufriadis et al. 2014) |
| DNAAF3 | 19q13.4 | *614566 | Assembly of axonemal IDA and ODA anddynein complexes | Absence of ODA and IDA | 10 | (Mitchison et al. 2012) |
| C21orf59 | 21q22.1 | *615494 | DA assembly | Absence of both ODA and IDA components | 4 | (Austin-Tse et al. 2013) |
| CCDC103 | 17q21.31 | *614677 | Fundamental factor for DA binding to cilia MT | Partial loss of ODA complexes | 10 | (Panizzi et al. 2012) |
| CCDC114 | 19q13.33 | *615038 | Component of the ODA docking complex | Absence of ODAs | 23 | (Knowles et al., 2013a; Onoufriadis et al., 2013) |
| DRC1 | 2p23.3 | *615288 | Regulation of the dynein motors | Severe defects in assembly of the N-DRC | 4 | (Wirschell et al. 2013) |
| CCDC39 | 3q26.33 | *613798 | Assembly of DRC and IDA complexes | Displacement of outer doublets, reductions of IDA and abnormal radial spokes and nexin links | 59 | (Merveille et al. 2011; Antony et al. 2013) |
| CCDC40 | 17q25.3 | *613799 | Assembly of DRC and IDA complexes | Misplacement of the central pair of MT and defective assembly of IDA and DRC | 54 | (Merveille et al. 2011; Antony et al. 2013) |
| CCDC65 | 12q13.12 | *611088 | Assembly of the N-DRC | Normal axonemal ultrastructure, only with a reduction in IDA and nexin links. | 4 | (Austin-Tse et al., 2013; Horani et al., 2013a) |
| DNAAF1 (*LRRC50) | 16q24.1 | *613190 | Pre-assembly and/or targeting of dynein-arm complexes | Marked reduction of both ODA and IDA | 8 | (Loges et al. 2009; Duquesnoy et al. 2009) |
| DNAAF2 (KTU) | 14q21.3 | *612517 | Pre-assembly of dynein arm complexes | Absence or defects of ODA and IDA | 3 | (Omran et al. 2008) |
| DNAH11 | 7p21 | *603339 | Encodes a ciliary ODA protein | Normal axonemal ultrastructure | 24 | (Bartoloni et al. 2002; Lucas et al. 2012; Knowles et al. 2012; Schwabe et al. 2008) |
| DNAH5 | 5p15.2 | *603335 | Important for function of the ODA complex. | Absence of ODAs | 93 | (Djakow et al., 2012; Failly et al., 2009; Hornef et al., 2006; Knowles et al., 2013a; Olbrich et al., 2002) |
| DNAI1 | 9p13.3 | *604366 | 43 | (Failly et al., 2008; Guichard et al., 2001; Pennarun et al., 1999; Zariwala et al., 2006; Ziętkiewicz et al., 2010) |
DA- Dynein Arms; ODA- Outer Dynein Arms; IDA- Inner Dynein Arms; DRC- Dynein Regulatory Complex; N-DRC- Nexin-Dynein Regulatory Complex; MT-Microtubules; N.D. Not-determined. LC- Light Chain; HC- Heavy Chain;
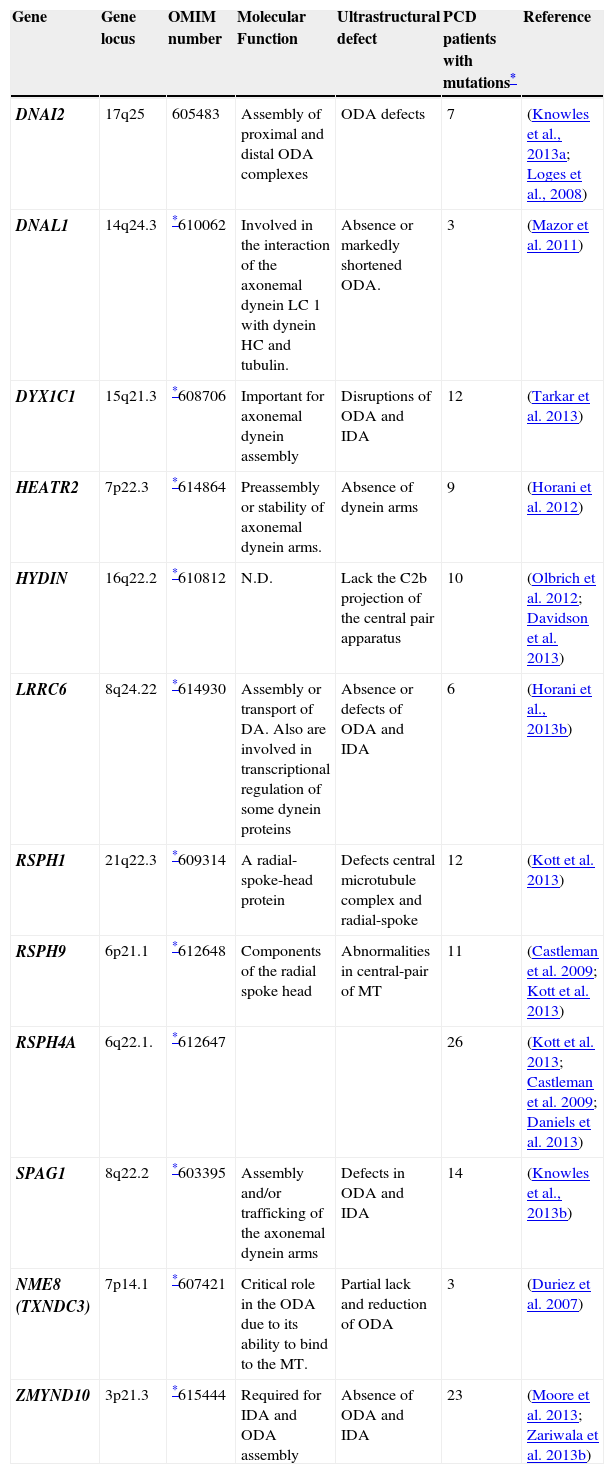
(continued) List of genes known to be associated with Primary Ciliary Dyskinesia (PCD) and main ultrastructural defects found in the axoneme of PCD patients.
| Gene | Gene locus | OMIM number | Molecular Function | Ultrastructural defect | PCD patients with mutations* | Reference |
|---|---|---|---|---|---|---|
| DNAI2 | 17q25 | 605483 | Assembly of proximal and distal ODA complexes | ODA defects | 7 | (Knowles et al., 2013a; Loges et al., 2008) |
| DNAL1 | 14q24.3 | *610062 | Involved in the interaction of the axonemal dynein LC 1 with dynein HC and tubulin. | Absence or markedly shortened ODA. | 3 | (Mazor et al. 2011) |
| DYX1C1 | 15q21.3 | *608706 | Important for axonemal dynein assembly | Disruptions of ODA and IDA | 12 | (Tarkar et al. 2013) |
| HEATR2 | 7p22.3 | *614864 | Preassembly or stability of axonemal dynein arms. | Absence of dynein arms | 9 | (Horani et al. 2012) |
| HYDIN | 16q22.2 | *610812 | N.D. | Lack the C2b projection of the central pair apparatus | 10 | (Olbrich et al. 2012; Davidson et al. 2013) |
| LRRC6 | 8q24.22 | *614930 | Assembly or transport of DA. Also are involved in transcriptional regulation of some dynein proteins | Absence or defects of ODA and IDA | 6 | (Horani et al., 2013b) |
| RSPH1 | 21q22.3 | *609314 | A radial-spoke-head protein | Defects central microtubule complex and radial-spoke | 12 | (Kott et al. 2013) |
| RSPH9 | 6p21.1 | *612648 | Components of the radial spoke head | Abnormalities in central-pair of MT | 11 | (Castleman et al. 2009; Kott et al. 2013) |
| RSPH4A | 6q22.1. | *612647 | 26 | (Kott et al. 2013; Castleman et al. 2009; Daniels et al. 2013) | ||
| SPAG1 | 8q22.2 | *603395 | Assembly and/or trafficking of the axonemal dynein arms | Defects in ODA and IDA | 14 | (Knowles et al., 2013b) |
| NME8 (TXNDC3) | 7p14.1 | *607421 | Critical role in the ODA due to its ability to bind to the MT. | Partial lack and reduction of ODA | 3 | (Duriez et al. 2007) |
| ZMYND10 | 3p21.3 | *615444 | Required for IDA and ODA assembly | Absence of ODA and IDA | 23 | (Moore et al. 2013; Zariwala et al. 2013b) |
DA- Dynein Arms; ODA- Outer Dynein Arms; IDA- Inner Dynein Arms; DRC- Dynein Regulatory Complex; N-DRC- Nexin-Dynein Regulatory Complex; MT-Microtubules; N.D. Not-determined. LC- Light Chain; HC- Heavy Chain;
The first gene in which mutations were found to be associated with PCD was DNAI151 that is an axonemal dynein IC gene, found in the ODA. It is localized on chrmosome 9p13-p21 and is composed of 20 exons encoding a protein with 699 amino acids. Mutations of DNAI1 have been identified in patients with PCD/KS, with often, but not always, absent or shortened ODA.51-53 This gene has been one of the most studied, although a relativly low prevalence of this disease was described (about 10% of PCD patients).54,55 The gene DNAH5, localized at chromosome 5p15.2, encodes a HC of the ODA and comprises 79 exons. Defects of the ODA were found associated with DNAH5 mutations in patients with PCD. Mutations in the DNAH5 gene are responsible for approximately 15–24% of all PCD cases.56,57 Overall, these data suggest that DNAI1 and DNAH5 genes are important for the function of the ODA complex and previous studies suggested that mutations in these genes are a major cause of PCD, given that they account for up to 38% of all patients.44,49,55
The genes CCDC39 and CCD40 are also of great importance in PCD, as they express integral components of the dynein regulatory complex. The human CCDC39 gene is localized at chromosome 3q26.33 and encodes a 941-amino acid protein that was shown to be essential for the assembly of the IAD and of the DRC, since mutations in CCDC39 result in failure to correctly assemble IDA complexes, DRC and radial spokes. This causes disorganization of the Ax, including mislocalized peripheral doublets, displacement, absence or supernumerary central pair and dyskinetic beating.58 The CCDC40 gene (localized in chromosome 17q25.3) contains 20 exons and encodes for CCDC40 protein with 1,142 amino acids. Mutations in CCDC40 were found in subjects with PCD, and ultrastructural analyses showed defects in several Ax structures, including disorganization of the MT doublets, absent or shifted central pairs, reduction in the mean number or absence of IDA, and abnormal radial spokes and nexin links. Nevertheless the ODA appeared normal.59 The, CCDC40 protein appears to be required for Ax recruitment of CCDC39, and both proteins interact with N-DRC (nexin) components, playing a role in IDA attachment.58,59 A recent study detected mutations in both genes CCDC39 and CCDC40 among 69% of individuals with PCD, with ultrastuctural defects that are indistinguishable at electronic microcope.60
Dysplasia of the Fibrous SheathDysplasia of the Fibrous Sheath (DFS), also called stump tail syndrome, is one of the most severe abnormalities of the sperm flagellum and causes extreme ATZ.32,34 Marked hyperplasia and disorganization of the FS is the typical diagnostic finding in these cases. In addition, the majority of sperm from affected individuals have short, thick, irregular flagella with no clear distinctions among the midpiece, principal piece and end piece (Figs. 2D and 2E). It is also observed partial or total lack of dynein arms, absence of the central pair (in about half of the cases), absence of a normal An and disassemble of the mitochondrial sheath.33,34,61,62 Although only occasionally associated with lack of IDA/ODA, DFS is considered a variant of PCD.
In humans, A-kinase anchoring proteins-3 and 4 (AKAP3, AKAP4) are the most abundant structural proteins, anchoring cyclic adenosine monophosphate–dependent-protein-kinase-A to the FS.25 The AKAP4 gene, localized in chromosome Xp11.22, is expressed in the post-meiotic phase of spermatogenesis and encodes an AKAP4 protein, with 854 amino-acids, that is restricted to the PP of the flagellum.63 AKAP4 play a major role in completing FS assembly, and thus in sperm motility.25,63AKAP3 gene, located at chromosome 12p13.3, encodes another of the major proteins of the FS. The ~110-kDa AKAP3 protein, with 853 amino-acids, is synthesized in round spermatids, incorporated into the FS simultaneously with the formation of rib precursors. AKAP3 is involved in organizing the basic structure of the FS.63,64 Although previous reports suggested that mutations in AKAP3 and AKAP4 genes are the genetic cause of the DFS phenotype,32,63,65-67 no strong evidences are yet available for the involvement of specific genes in the pathogenesis of DFS.34,68
Final remarksThe molecular components that were referred in this review are merely a small portion of all molecular components and interactions that exist in the complex sperm flagellum. There is still a long journey to make in order to fully understand all the genetics of sperm flagellum and the molecular components that are responsible for the assembly of the sperm flagellum. The automated Sanger sequencing method has dominated genetics for the last two decades and still gives huge contributes to the scientific knowledge about many genetic disorders. However, the limitations of automated Sanger sequencing, such as high cost and low throughput, propelled the need for new sequencing technologies. The next-generation sequencing (NGS) technologies, such as whole the genome sequencing (WGS), are revolutionizing genetics and the scientific/medical research. They are able to produce an enormous amount of data in a cheaper and faster way.69 Nevertheless, the enormous quantity of data provided is difficult to handle and analyse.
Exome sequencing (ES) is an efficient strategy to selectively sequence the coding regions of the genome (exome) being an alternative to WGS. With ES, the amount of data is reduced, as well as, the costs with an estimated 10 to 20-fold reduction in raw sequencing data needed as compared to WGS.70 It is believed that the exome contains the great majority of the disease-causing mutations of all genome, and consequently ES is described as a powerful discovery tool. It has already contributed to the identification of new genes involved in PCD71-75 and it will certainly help to increase our knowledge about the genetic causes of sperm immotility and infertility.
Conflicts of interestsThe authors state that they have no conflict of interests.Aknowledgements
