


El presente estudio describe la implementación de un test de cribado de portadores de enfermedades genéticas autosómicas recesivas y enfermedades ligadas al cromosoma X en un programa de donación de ovocitos (donantes de ovocitos y parejas masculinas de las receptoras). El test empleado basado en tecnología Next-Generation Sequencing (NGS), cubría 200 genes (68 mediante análisis completo de secuencia codificante y 132 por estrategia dirigida de análisis) asociados a 314 enfermedades (277 enfermedades autosómicas recesivas y 22 ligadas al cromosoma X). El resultado obtenido tras 2,5 años mostró un alto grado de aceptación (implementación>80%). Se identificaron un 56,4% de individuos (761/1.350) portadores de al menos una mutación patogénica. Asimismo se identificó que el 1,9% de las candidatas a donantes eran portadoras de una enfermedad ligada al cromosoma X por lo que se excluyeron del programa de Donación de Ovocitos. La carga mutacional media fue de 0,84 mutaciones por muestra. Se identificaron un 3,4% de preasignaciones donante-receptor con alto riesgo reproductivo para alguna enfermedad genética recesiva (fibrosis quística, hiperplasia suprarrenal congénita, sordera congénita no-sindrómica, alfa-talasemia, fiebre mediterránea familiar, enfermedad de Niemann-Pick, dishormonogénesis tiroidea tipo 6 e hipoplasia de cartílago-pelo). La asignación definitiva se realizó teniendo en cuenta los resultados de los estudios genéticos. El estado de portador heterocigoto para una enfermedad autosómica recesiva no fue motivo de exclusión del programa de Donación de Ovocitos, pero implicó la selección de una receptora cuya pareja masculina no fuera portadora de la misma enfermedad. El asesoramiento genético de donantes y receptores en las diferentes etapas del proceso clínico de la donación de ovocitos fue esencial para disminuir la ansiedad, conseguir una buena comprensión de los resultados y poder otorgar las recomendaciones pertinentes sobre el riesgo reproductivo propio o de sus familiares, cuando fue necesario.
The present study describes the implementation of a carrier screening test for autosomal recessive and X-linked diseases in an Oocyte Donation Programme (donors and male partners of the recipients). The Next-Generation Sequencing (NGS) based test covered 200 genes (68 genes by complete sequencing of the coding region and 132 sequenced by a targeted mutation analysis) associated with 314 diseases (277 recessive autosomal diseases and 22 X-linked). The results obtained after 2.5 years showed a high degree of acceptance (implementation rate>80%). Among participants, 56.4% (761/1350) were identified as carriers of at least one gene mutation. It was also identified that 1.9% of donor candidates were carriers of X-linked diseases, and for this reason were excluded from the Oocyte Donation programme. The mean carrier burden was 0.84 mutations per sample. There were 3.4% of preassigned donor-recipient matches with a high reproductive risk for transmitting a severe autosomal-recessive genetic condition to their offspring (cystic fibrosis, classical congenital adrenal hyperplasia, autosomal-recessive non-syndromic sensorineural deafness, alpha thalassemia, familial Mediterranean fever, Niemann-Pick disease, thyroid dyshormogenesis type 6 and cartilage-hair hypoplasia). The definitive matching was made taking into account the genetic results. A carrier state for an autosomal recessive condition was not an exclusion criterion for the Oocyte Donation programme, although it implied the assignation of the donor to a recipient whose male partner was not a carrier for the same recessive condition. Genetic counselling at different stages of the clinical process was essential in order to achieve the purposes of decreasing anxiety in the gamete donors and recipient couples, to achieve a high understanding of the results, and to give appropriate recommendations about their own reproductive risk or that of their relatives, when necessary.
La donación de ovocitos (DO) es una de las técnicas de reproducción asistida más utilizada en la actualidad. Anualmente se realizan en España más de 16.000 ciclos con DO frescos o criopreservados, representando el 14,3% de los ciclos de técnicas de reproducción asistida realizados y la tasa de gestación por ciclo de recepción supera el 50% (SEF, 2014). Los centros de fertilidad autorizados tienen la responsabilidad de velar no solo por la salud de sus pacientes y donantes, sino también por la de los niños nacidos fruto de la donación de gametos. En este sentido, el asesoramiento preconcepcional y la evaluación del riesgo genético tienen un papel crucial en la prevención de enfermedades hereditarias en la descendencia.
El Real Decreto-ley 9/2014 por el que se establecen las normas de calidad y seguridad para la donación en España exige el cumplimiento de un protocolo de estudio en los donantes en el que se expone la necesidad de evaluar el riesgo de transmisión de enfermedades hereditarias conocidas y presentes en la familia de los donantes e indica que se debe evaluar la carga genética con relación a la existencia de genes autosómicos recesivos en base al conocimiento científico y a la prevalencia conocida en la etnia del donante. No obstante, la norma no especifica cuáles deben ser las pruebas concretas a realizar y tampoco define cuál es el punto de corte para considerar que una prevalencia sea suficientemente elevada para condicionar el test genético que se debe realizar al donante. Debido a la falta de concreción de la norma, se puede interpretar que la realización de estudios genéticos queda a criterio facultativo. En consecuencia, la Sociedad Española de Fertilidad (SEF) y la Asociación para el Estudio de la Biología de la Reproducción (ASEBIR) elaboraron un documento de consenso para interpretar y establecer unos criterios de mínimos para el estudio de los donantes (Alonso et al., 2012).
La mayoría de los centros de reproducción asistida en España, basándose en las recomendaciones de la SEF y ASEBIR, incluyen en el estudio de los donantes el cariotipo, el cribado mutacional dirigido del gen CFTR, el estudio de la premutación X-frágil y otras enfermedades según la etnia del/la donante, como la alfa/beta-talasemias para el área mediterránea, o la enfermedad de Tay-Sachs en los judíos Asquenazíes.
La aparición de las nuevas tecnologías genómicas y el creciente abaratamiento de estas han permitido ampliar el cribado de variantes genéticas a un elevado número de enfermedades. Inicialmente, aparecieron las tecnologías de genotipado que interrogan un número concreto de mutaciones puntuales o pequeñas inserciones y deleciones en los genes seleccionados de modo que solamente permiten explorar las mutaciones conocidas causantes de enfermedad a criterio del laboratorio en el momento de su diseño. Más recientemente, las nuevas tecnologías de secuenciación masiva (Next Generation Sequencing, NGS) permiten explorar de forma simultánea y sin sesgos casi cualquier mutación presente en toda la región codificante de genes asociados a enfermedad, sea o no conocida a priori. Esta revolución tecnológica ha impulsado el desarrollo de múltiples test genéticos, disponibles en el mercado desde el año 2012 (Lazarin et al., 2013). Algunos centros de reproducción asistida han empezado a implementarlo en la práctica clínica especialmente en los programas de donación de gametos (Martin et al., 2015; Abulí et al., 2016).
Debido a la gran diversidad de test genéticos disponibles con distinto número y tipo de enfermedades, y la ausencia de un consenso para establecer unos criterios para el cribado de enfermedades, en los últimos años diferentes sociedades científicas internacionales se han posicionado al respecto. Concretamente, el American College of Medical Genetics and Genomics (ACMG) conjuntamente con otras sociedades científicas norteamericanas y la European Society of Human Genetics (ESHG) publicaron individualmente sus posicionamientos en los que establecen los criterios para definir qué enfermedades deberían incluirse en un cribado ampliado de portadores (Grody et al., 2013; Edwards et al., 2015; Henneman et al., 2016). Los autores recomiendan que las enfermedades incluidas deben ser graves y accionables a nivel reproductivo. El cribado de enfermedades de aparición en edad adulta o con expresividad variable, penetrancia incompleta y fenotipos leves debería contemplarse de forma individual y solicitar consentimiento informado específico para este tipo de enfermedades.
En el contexto de la medicina reproductiva, en el año 2014, la European Society of Human Reproduction and Embryology (ESHRE) publicó un documento sobre los aspectos éticos del cribado genético en donantes de gametos (Dondorp et al., 2014). Los autores consideran que el cribado ampliado de portadores debería ser considerado en la práctica clínica, valorando términos de eficacia y proporcionalidad. El documento refleja la importancia de un consentimiento informado y un adecuado asesoramiento genético pre- y postest que garantice la interpretación de la patogenicidad de las variantes halladas y proporcione una información adecuada sobre las implicaciones de las mismas; así como la necesidad de salvaguardar los aspectos éticos fundamentales de cara a pacientes, donantes de gametos y familiares a riesgo. ASEBIR dio a conocer recientemente su posicionamiento sobre los estudios genéticos de cribado ampliado de enfermedades recesivas en los programas de donación. Este documento recomienda que los pacientes deben ser informados de la existencia de estos estudios genéticos teniendo en cuenta el estado del conocimiento y desarrollo tecnológico actual y, valorar en cada caso, sin obligatoriedad, su realización tanto al donante como a la pareja receptora (Boada et al., 2017).
En el presente estudio se reporta la experiencia clínica en la implementación de un test de cribado ampliado de portadores en un programa de DO con el fin de reducir el riesgo de transmisión de enfermedades genéticas hereditarias a la descendencia y los resultados obtenidos durante los 2 primeros años de experiencia.
Material y métodosSe realizó un estudio observacional retrospectivo descriptivo de 766 ciclos de DO realizados durante el periodo de enero 2014 a junio 2016.
PacientesEl estudio incluyó a un total de 1.389 individuos (623 candidatas a donante de ovocitos y un total de 766 parejas masculinas de receptoras de ovocitos) atendidos en el contexto de un programa de DO en el Departamento de Obstetricia, Ginecología y Reproducción de Salud de la Mujer Dexeus en Barcelona.
Selección de genesLa selección de genes incluidos en el panel de cribado ampliado de portadores se realizó siguiendo las recomendaciones de las sociedades científicas profesionales de la American College of Obstetricians and Gynecologists (ACOG) y el ACMG (Edwards et al., 2015).
La prueba cubrió 200 genes mórbidos OMIM (Online Mendelian Inheritance in Man, www.ncbi.nlm.nih.gov/omim) asociados a 314 enfermedades (277 enfermedades autosómicas recesivas y 22 ligadas al cromosoma X (tabla S1).
Diseño del panelEl test fue desarrollado mediante tecnología de NGS permitiendo la caracterización de mutaciones puntuales, inserciones, deleciones, variantes de número de copia y reordenamientos. Las regiones de interés fueron: i) todos los exones codificantes de genes asociados con enfermedades comunes en nuestra área geográfica; ii) estudio dirigido de mutaciones patogénicas conocidas en genes asociados con enfermedades raras.
De los 200 genes analizados, 68 se estudiaron en análisis completo de secuencia codificante mientras que en los 132 restantes se utilizó una estrategia dirigida de análisis.
Las deleciones en los genes HBA1, HBA2 y SMN1 fueron validadas mediante la técnica Multiplex Ligation-dependent Probe Amplification (MLPA) empleando kits comerciales (MRC-Holland, Amsterdam, Holanda) y las expansiones del triplete (CGG) de la región promotora del gen FMR1 asociado a X-frágil, fueron analizadas mediante el kit comercial AmplideX PCR/CE (Asuragen).
Protocolo de selección de donantesLas donantes se cribaron siguiendo el protocolo del centro (Martínez et al., 2008). El protocolo utilizado cumple los requisitos clínicos y legales que determina la normativa vigente (Real Decreto-ley 9/2014) respetando la condición de anonimato y los límites de edad de las donantes que establece la Ley 14/2006.
En nuestro programa de DO únicamente son aceptadas las candidatas con un recuento folicular antral>10 folículos, índice de masa corporal<28kg/m2, sin antecedentes personales o familiares de enfermedades genéticas hereditarias y estudio citogenético normal. Finalmente, el cribado genético de enfermedades hereditarias mendelianas en las donantes se realiza mediante el test qCarrier (qGenomics).
Protocolo de implementación del test qCarrierEl cribado ampliado de portadores se ofreció a todas las parejas receptoras de ovocitos y a las candidatas a donantes de ovocitos. La implementación del test de cribado ampliado de portadores se llevó a cabo gracias a una estrecha colaboración entre el Servicio de Medicina de la Reproducción y la Unidad de Medicina Genómica. La importancia de esta última se considera crucial para que los pacientes y donantes que se someten al test reciban un adecuado asesoramiento genético antes y después de la realización de la prueba por parte de un profesional especialista en genética.
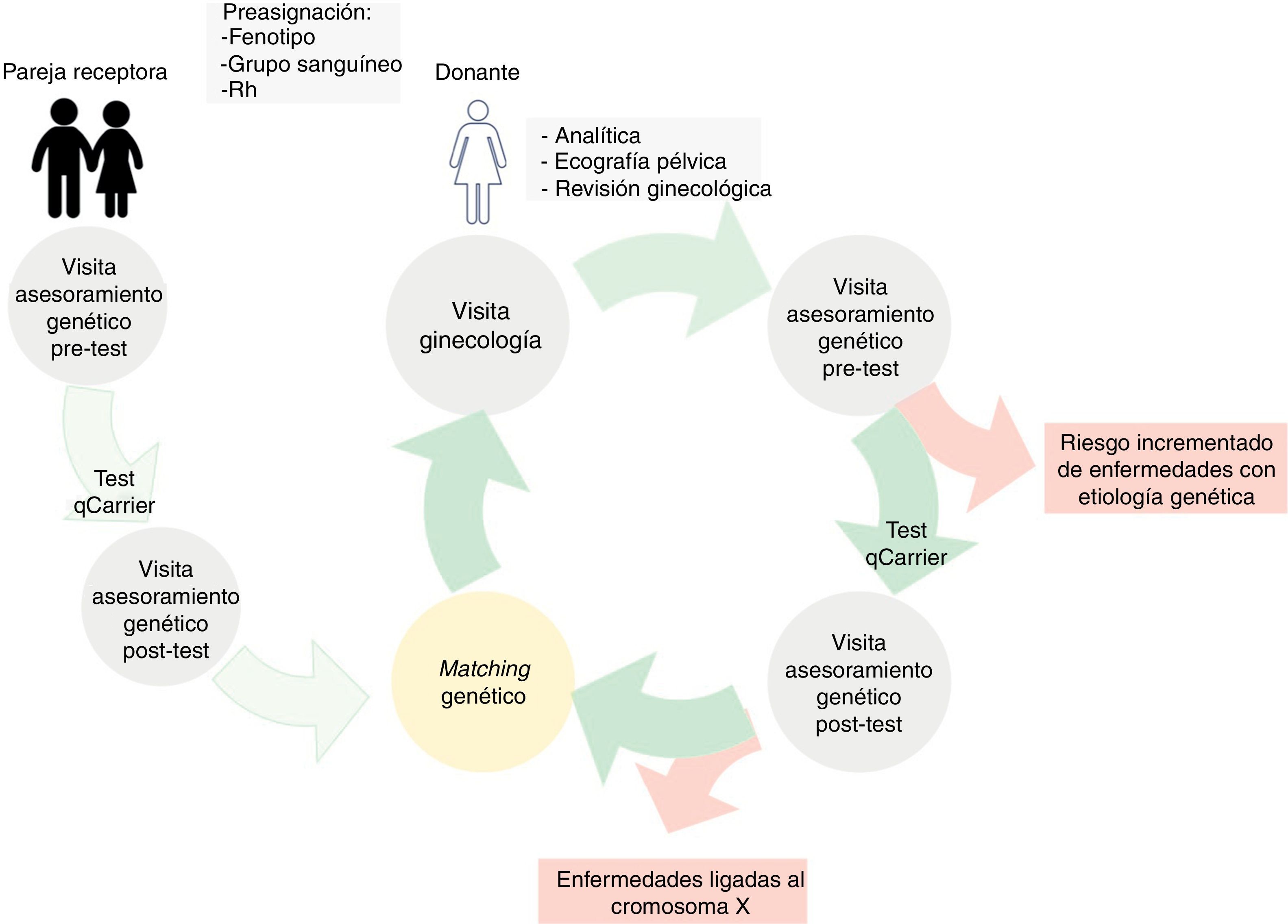
La preasignación donante-receptora se efectuó siguiendo los criterios de similitud fenotípica, grupo sanguíneo y factor Rh (fig. 1). Las donantes tienen una primera visita de asesoramiento genético en la Unidad de Medicina Genómica para recoger la historia familiar; aquellas con antecedentes de enfermedades genéticas hereditarias con alto riesgo de transmisión son descartadas del programa de donación. En esta visita pretest se explica en qué consiste el test de cribado ampliado de portadores, el tipo de enfermedades que estudia, los modelos de herencia y las implicaciones reproductivas que podrían tener estos resultados para ellas y sus familias. Del mismo modo, las parejas receptoras también reciben una visita de asesoramiento genético pretest. Las donantes y las parejas receptoras firman el consentimiento informado en el que establecen si desean o no conocer los resultados del test, incluyendo variantes de significado incierto (VUS) y/o hallazgos incidentales. Posteriormente se cursa el test de cribado ampliado de portadores a la pareja masculina de la receptora de ovocitos y a la candidata a donante de ovocitos preasignada. Por lo general, se obtiene una muestra de sangre para realizar el estudio genético, aunque aquellas parejas receptoras que residen en el extranjero también tienen la posibilidad de enviar una muestra de saliva tras una visita de asesoramiento genético mediante videoconferencia. La asignación definitiva de donante-receptora se realiza tras los resultados del estudio genético (fig. 1).

Para el proceso de asignación se consideran las mutaciones patogénicas, las VUS y las regiones génicas insuficientemente cubiertas en cada caso. Se descartan del programa de DO todas las candidatas portadoras de una enfermedad ligada al cromosoma X con alto riesgo reproductivo (fig. 1). El estado de portador heterocigoto para una enfermedad autosómica recesiva no es motivo de exclusión del programa de DO, pero implica la selección de una receptora cuya pareja masculina tenga un riesgo mínimo de ser portador de una mutación para la misma enfermedad.
Las donantes y la pareja receptora de ovocitos son visitadas en la Unidad de Medicina Genómica para la entrega de resultados. En esta visita de asesoramiento genético postest se explica el estado o no de portador, el fenotipo asociado, el riesgo de descendencia afecta y las recomendaciones reproductivas para él/ella y su familia, y se emite un informe en cada caso. Las donantes y la pareja receptora reciben un informe de asesoramiento genético que incluye su propio riesgo reproductivo y las recomendaciones futuras para él/ella, su pareja y otros miembros de la familia.
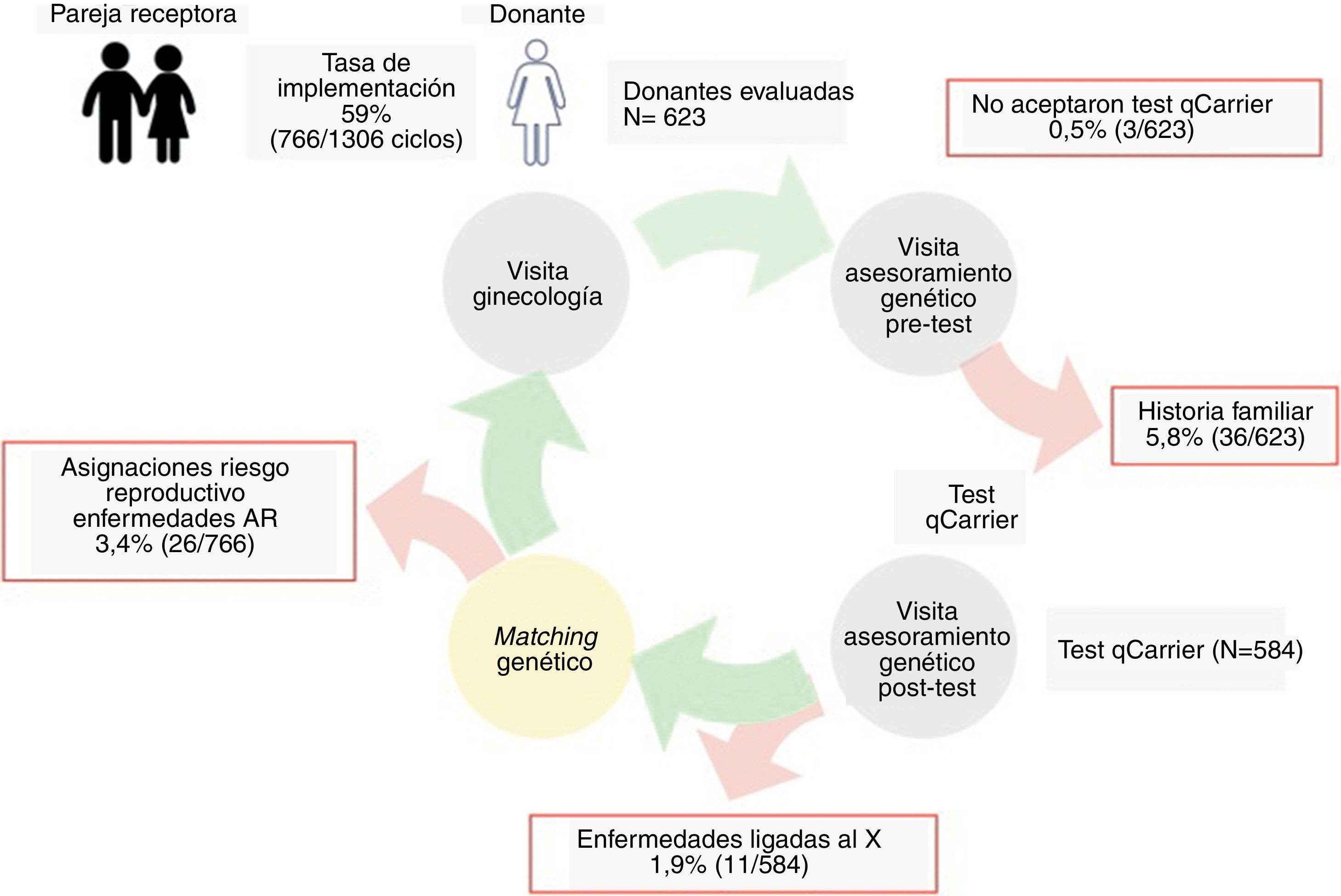
ResultadosEl estudio incluyó a un total de 1.389 individuos procedentes de nuestro programa de DO (623 candidatas a donante de ovocitos y 766 parejas masculinas de receptoras de ovocitos). De las 623 candidatas a donante evaluadas, el 0,5% (3/623) no aceptaron realizar el cribado ampliado de portadores y el 5,8% (36/623) fueron descartadas por historia familiar (fig. 2).

El cribado ampliado de portadores se realizó a un total de 1.350 pacientes: 584 candidatas a donantes de ovocitos (43,3%) y un total de 766 parejas masculinas de receptoras de ovocitos (56,7%).
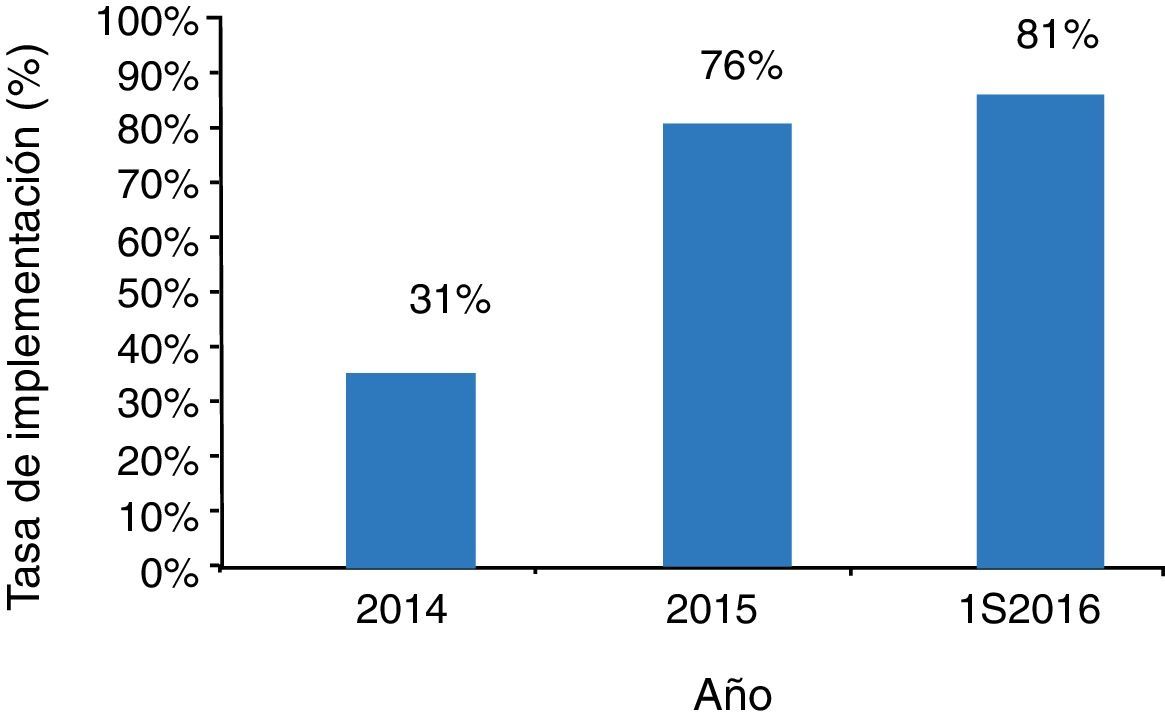
La tasa de implementación clínica del cribado ampliado de portadores en nuestro programa de DO durante el primer semestre del año 2016 fue del 81% (325/403). Esta cifra ha ido en aumento desde su inicio, hasta alcanzar el valor actual (fig. 3). La tasa media de implementación considerando todo el periodo de estudio fue del 59% (766/1306).
 en el programa de donación de ovocitos de Salud de la Mujer Dexeus.")
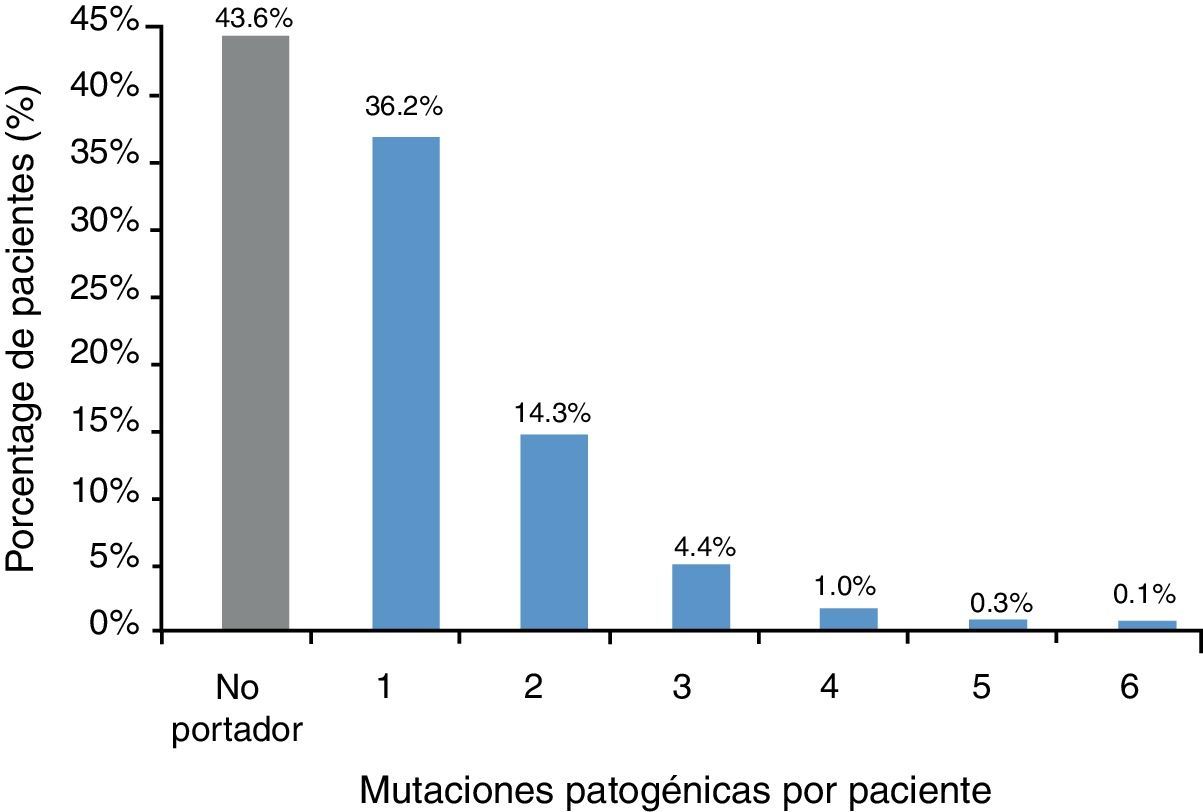
La implementación del test qCarrier en el contexto clínico ha permitido identificar a un 56,4% de individuos (761/1350) portadores de al menos una mutación patogénica o probablemente patogénica (variante tipo nonsense, splicing o frameshift) en un gen causante de una enfermedad genética mendeliana. La mayoría de los portadores presentaban una mutación (un 36,2% del total de individuos) y un 20,2% de individuos eran portadores de 2 (14,3%) o más (5,9%) enfermedades genéticas (fig. 4).

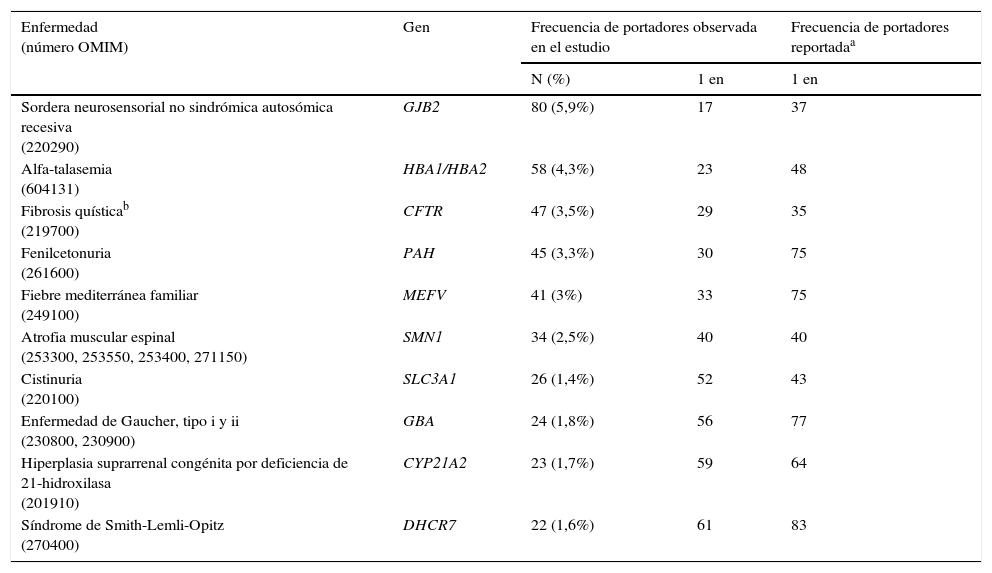
La carga mutacional media fue de 0,84 mutaciones por muestra. La tabla 1 muestra las 10 enfermedades genéticas más frecuentes detectadas en nuestra población de estudio incluyendo las frecuencias de portador previamente reportadas en la literatura (Lazarin et al., 2013). La tabla S2 muestra la lista completa de genes en los cuales se han detectado mutaciones patogénicas.
Listado de las 10 enfermedades genéticas más frecuentes detectadas mediante el test qCarrier en nuestra población
| Enfermedad (número OMIM) | Gen | Frecuencia de portadores observada en el estudio | Frecuencia de portadores reportadaa | |
|---|---|---|---|---|
| N (%) | 1 en | 1 en | ||
| Sordera neurosensorial no sindrómica autosómica recesiva (220290) | GJB2 | 80 (5,9%) | 17 | 37 |
| Alfa-talasemia (604131) | HBA1/HBA2 | 58 (4,3%) | 23 | 48 |
| Fibrosis quísticab (219700) | CFTR | 47 (3,5%) | 29 | 35 |
| Fenilcetonuria (261600) | PAH | 45 (3,3%) | 30 | 75 |
| Fiebre mediterránea familiar (249100) | MEFV | 41 (3%) | 33 | 75 |
| Atrofia muscular espinal (253300, 253550, 253400, 271150) | SMN1 | 34 (2,5%) | 40 | 40 |
| Cistinuria (220100) | SLC3A1 | 26 (1,4%) | 52 | 43 |
| Enfermedad de Gaucher, tipo i y ii (230800, 230900) | GBA | 24 (1,8%) | 56 | 77 |
| Hiperplasia suprarrenal congénita por deficiencia de 21-hidroxilasa (201910) | CYP21A2 | 23 (1,7%) | 59 | 64 |
| Síndrome de Smith-Lemli-Opitz (270400) | DHCR7 | 22 (1,6%) | 61 | 83 |
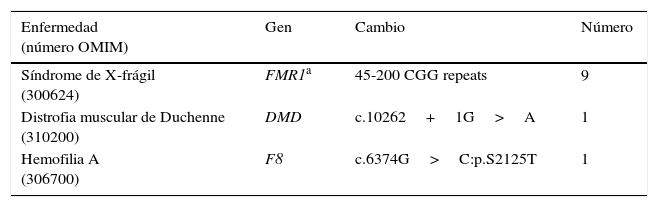
Se han descartado 1,9% (11/584) candidatas a donante de ovocitos tras resultar portadoras de una enfermedad ligada al cromosoma X incluyendo la hemofilia A, los alelos intermedios o con la premutación X-frágil y la distrofia muscular de Duchenne (tabla 2).
Donantes de ovocitos descartadas por mutaciones en genes causantes de enfermedades ligadas al cromosoma X
| Enfermedad (número OMIM) | Gen | Cambio | Número |
|---|---|---|---|
| Síndrome de X-frágil (300624) | FMR1a | 45-200 CGG repeats | 9 |
| Distrofia muscular de Duchenne (310200) | DMD | c.10262+1G>A | 1 |
| Hemofilia A (306700) | F8 | c.6374G>C:p.S2125T | 1 |
Se han identificado un 3,4% (26/766) de asignaciones con alto riesgo reproductivo para enfermedades recesivas, es decir que la donante preasignada y la pareja masculina eran portadores de mutaciones en el mismo gen (fig. 2). Estos casos se han resuelto realizando la asignación de una nueva donante no portadora de mutaciones en el mismo gen(es). Concretamente se han identificado asignaciones con riesgo de descendencia afecta de fibrosis quística (formas clásicas y fenotipos relacionados con el gen CFTR), hiperplasia suprarrenal congénita, sordera congénita no sindrómica, alfa-talasemia, fiebre mediterránea familiar, enfermedad de Niemann-Pick, dishormonogénesis tiroidea tipo 6 e hipoplasia de cartílago-pelo.
DiscusiónEl presente estudio reporta la experiencia en la incorporación de un test de cribado ampliado de portadores en un programa de DO, con una tasa de implementación clínica prácticamente generalizada durante el último periodo de estudio (81%). La incorporación del test genético ha permitido reducir el riesgo de transmisión de enfermedades genéticas a los niños nacidos a través de DO. Se ha identificado un 3,4% de asignaciones con alto riesgo reproductivo de enfermedades recesivas y se ha descartado el 1,9% de donantes portadoras de mutaciones ligadas al cromosoma X.
En nuestro conocimiento, tasas tan elevadas de implementación de un test de cribado ampliado de portadores en un programa de DO no han sido reportadas hasta la fecha. La tasa de implementación se ha incrementado desde el 30% en el primer año al 81% en el primer semestre del pasado año. Esto representa un alto grado de implementación y aceptación por parte de los pacientes, fruto de una buena labor de información. Según nuestra experiencia, los principales motivos por los que algunas parejas deciden no realizar el cribado ampliado de portadores son razones económicas o limitaciones geográficas, a pesar de que se ofrece la posibilidad de enviar muestras utilizando el kit de saliva.
El objetivo de un test de portadores es aumentar las probabilidades de detección de portadores de enfermedades genéticas para potenciar la autonomía reproductiva y facilitar la toma de decisiones informada, al tiempo que disminuir el número de embarazos de niños afectos de enfermedades genéticas recesivas. Nuestro protocolo en el programa de DO incluye la revisión de la historia personal y familiar por un profesional en genética, por lo que es posible identificar cualquier antecedente familiar de enfermedad hereditaria dominante o con alta susceptibilidad genética. En gran parte por este motivo, los genes asociados a estos fenotipos no están incluidos en el test genético, aunque técnicamente también podrían ser analizados. Actualmente, el objetivo del cribado ampliado de portadores se centra en la identificación de portadores heterocigotos de condiciones genéticas, recesivas y ligadas al cromosoma X en su mayor parte, de afectación grave en edad temprana que pueden pasar desapercibidas en una historia familiar.
Mediante el test qCarrier se han identificado donantes portadoras de mutaciones ligadas al cromosoma X (1,9%) y se ha identificado asignaciones con alto riesgo reproductivo de enfermedades recesivas (3,4%). Esta cifra podría estar sobreestimada si tenemos en cuenta la frecuencia reportada de parejas portadoras de la misma enfermedad genética grave (1-2%) (Ropers, 2012), lo que podría ser explicado por nuestro tamaño muestral y porque las asignaciones con alto riesgo reproductivo incluyen variantes con alta frecuencia poblacional, como es el alelo polimórfico 5T del gen CFTR, asociadas a fenotipos no clásicos causantes de formas leves de la enfermedad. Respecto a las donantes descartadas por ser portadoras de mutaciones ligadas al cromosoma X, cabe mencionar que este alto porcentaje incluye los alelos intermedios del gen FMR1. Los resultados reportados en el artículo publicado por Martin et al. en 2015, en el que identificaron un 5% de asignaciones con alto riesgo y un 1,9% de donantes portadoras de mutaciones ligadas al cromosoma X (Martin et al., 2015), no son comparables debido a las diferencias de los paneles genéticos utilizados con diferente número de genes y diseño de estudio.
De acuerdo con las recomendaciones de las diferentes sociedades científicas, la implementación de un test genético de portadores debe desarrollarse en el contexto de una unidad de genética clínica para garantizar un adecuado asesoramiento genético antes y después de realizar el estudio con la finalidad de que las parejas y las donantes realicen una toma de decisiones informada, conozcan las implicaciones del resultado para ello/as y sus familiares, y para salvaguardar los aspectos éticos en donantes y pacientes. En base a nuestra experiencia consideramos importante destacar algunos aspectos relacionados con el asesoramiento genético.
En primer lugar es importante transmitir a los pacientes que todos los individuos somos portadores de enfermedades genéticas. Estudios recientes del exoma completo han revelado que cada individuo es portador de 2 a 8 mutaciones patogénicas (Gao et al., 2015; Lek et al., 2016). Esto ayuda tanto a las parejas receptoras como a las donantes a disminuir el riesgo de discriminación o estigmatización y a comprender que no existe un gameto libre de mutaciones.
Otro aspecto importante derivado de la aplicación de un cribado ampliado de portadores es evitar la «falsa tranquilidad». La reproducción humana conlleva un riesgo inherente que no podemos excluir completamente. No es posible descartar totalmente la existencia o presencia de mutaciones causantes de enfermedades genéticas. No es posible mediante los test disponibles comercialmente evitar las enfermedades genéticas debidas a mutaciones de novo, y normalmente, las pruebas de cribado incluyen un número limitado de genes y enfermedades. En consecuencia, los pacientes deben ser informados de que los estudios genéticos implementados en los programas de DO van dirigidos a reducir los riesgos para algunas enfermedades sin eliminarlos completamente. Además, a menudo los paneles dirigidos no cubren toda la secuenciación completa del gen, sino solo las mutaciones prevalentes en los exones codificantes. En este sentido, es importante que el asesoramiento genético incluya la comunicación de los riesgos residuales para las diferentes enfermedades estudiadas.
A pesar de tratarse de un estudio dirigido, también existe cierta probabilidad de que se produzcan hallazgos incidentales relacionados con las enfermedades objeto de estudio. Es importante informar que el estudio de portadores puede revelar un diagnóstico predictivo o presintomático puesto que el estado heterocigoto para algunas enfermedades puede revelar además un riesgo para una enfermedad de aparición tardía. Por ejemplo, el estado heterocigoto del gen ATM se asocia con un incremento del riesgo de cáncer de mama en mujeres, los portadores de enfermedad de Gaucher tienen un riesgo incrementado de desarrollar enfermedad de Parkinson y las portadoras de un alelo con premutación X-frágil presentan un riesgo de fallo ovárico prematuro y pueden desarrollar una enfermedad neurodegenerativa (FXTAS) en edad adulta. El conocimiento sobre el riesgo de enfermedad asociado a un determinado genotipo será mayor en un futuro, incrementando así la complejidad del asesoramiento genético en la implementación del cribado ampliado. Por todo ello, es importante realizar un asesoramiento genético pretest muy exhaustivo y obtener la conformidad del paciente o la donante mediante un consentimiento informado que incluya el derecho a saber y no saber este tipo de hallazgos.
Todavía existe un gran número de variantes sin clasificar a pesar de que se están efectuando grandes esfuerzos para la interpretación de variantes genéticas. En este sentido, las VUS representan un reto para el asesor genético de cara a su interpretación, manejo, y la ansiedad que puede derivarse de un resultado de significado clínico desconocido. Esto requiere un extenso asesoramiento genético previo al test que incluye comunicar la posibilidad de obtener este tipo de resultados inciertos que no van a cambiar las pautas de actuación clínica. Durante el periodo estudiado, se ha adoptado el protocolo de reportar las VUS al paciente a no ser que indique explícitamente que no desea conocer esta información.
La información brindada durante las sesiones de asesoramiento genético es incorporada satisfactoriamente en las parejas con deseo reproductivo. Sin embargo, la situación es diferente para la población de donantes de ovocitos. En primer lugar, la mayoría de los donantes no tienen un deseo reproductivo inmediato y no acuden a la clínica con la intención de conocer su riesgo genético reproductivo. Ello plantea una situación especial que debe ser considerada en las sesiones de asesoramiento genético. La mayoría de ellas entienden el concepto y las consecuencias reproductivas de ser portadoras y, en nuestra experiencia, solo una minoría opta por no ser informada acerca de los resultados del estudio.
Los principales argumentos en contra del cribado de enfermedades genéticas en medicina reproductiva son que sus resultados pueden llegar a ser perjudiciales desde un punto de vista psicoemocional para los donantes y sus familiares (Dondorp et al., 2014) y la preocupación de una posible estigmatización (Bream y Lott, 2010). En este sentido, nuestro grupo está llevando a cabo un estudio para evaluar el impacto emocional y psicológico de la implementación del test de cribado ampliado en donantes de ovocitos. Si bien los resultados preliminares reflejan niveles superiores de ansiedad y depresión en las donantes portadoras de alguna enfermedad en comparación con las donantes no portadoras, en ninguno de los casos superan el umbral patológico. Estos resultados indican que la implementación de un estudio genético de portadores en donantes de ovocitos con un adecuado asesoramiento genético no tiene un impacto emocional y psicológico significativo y parece ser bien aceptado por nuestra población de donantes (datos no publicados).
La comunicación del estado de portadora de la donante a las parejas receptoras es otro aspecto importante que ha generado controversia y que puede resultar en una demanda habitual en la práctica clínica. En este sentido, ASEBIR recomienda informar de que se dispone de la información del perfil genético y, si se solicita, podrá informarse de aquello que tenga transcendencia clínica para los receptores y su descendencia (Boada et al., 2017). Aunque el estado de portador no tiene implicaciones clínicas directas para la salud del individuo, sí puede ser una información útil para el futuro reproductivo de los hijos nacidos. Sin embargo, la utilidad real a largo plazo probablemente sea escasa ya que es notorio que el avance del conocimiento en el campo de la genómica permitirá, en un plazo de tiempo no muy lejano, disponer de la secuencia del genoma a nivel individual conociendo nuestro perfil genético completo.
Debido al avance entorno a la genómica, los test deben ser dinámicos, capaces de actualizarse en base al nuevo conocimiento y experiencias previas. Las nuevas versiones de los test de cribado ampliado de portadores deberían incluir genes y variantes a la luz de los nuevos conocimientos generados gracias a las técnicas de secuenciación masiva. Uno de los mayores problemas al que nos hemos enfrentado son los genes que no quedan totalmente cubiertos por lo que puede existir un riesgo residual considerable (Lazarin et al., 2013), no siempre aceptado por los pacientes. Esto puede plantear la realización de estudios moleculares adicionales del gen en la pareja o donante de un portador. Por lo tanto, para futuros paneles de cribado de portadores, la cobertura completa de estos genes debería ser una estrategia necesaria, ya incluida en nuevas versiones del test.
En resumen, el presente estudio describe la implementación de un test de cribado de portadores de enfermedades genéticas autosómicas recesivas y enfermedades ligadas al cromosoma X. El panel se ha aplicado en un programa de DO con un alto grado de implementación (>80%) para detectar el estado de portador de donantes de ovocitos y las parejas masculinas de las receptoras. El test permitió identificar y excluir del programa de DO a un 1,9% de las candidatas por ser portadoras de una enfermedad ligada al cromosoma X, y además identificar a un 3,4% de asignaciones donante-receptor con alto riesgo reproductivo para una de las enfermedades genéticas recesivas. El asesoramiento genético en las diferentes etapas del proceso clínico de la DO es esencial para disminuir la ansiedad de pacientes y donantes, así como para salvaguardar los principios éticos fundamentales de la práctica clínica.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que este estudio ha sido aprobado por la Cátedra de Investigación en Obstetricia y Ginecología del centro y que en todos los casos los pacientes y donantes han sido debidamente informados y asesorados genéticamente. Asimismo, declaran que para este trabajo de investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores BRS y LA son trabajadores de qGenomics, empresa dedicada a comercializar el test de portadores qCarrier.
Este trabajo ha sido realizado bajo los auspicios de la Cátedra de Investigación en Obstetricia y Ginecología (CIOG) del Hospital Universitario Dexeus.
