



Desde la descripción hace 5 décadas de la hidrocefalia crónica del adulto idiopática (HCAi), su fisiopatología ha sido considerada básicamente relacionada con el efecto que la dilatación ventricular ejerce sobre las estructuras adyacentes al sistema ventricular. Sin embargo, las alteraciones en el flujo sanguíneo cerebral (FSC) y, sobre todo, la reducción en el recambio licuoral parecen emerger como componentes fisiopatológicos principales de esta enfermedad.
DesarrolloEn la HCAi se observa una compresión del tracto piramidal, de los circuitos cortico-subcorticales fronto-estriatales y fronto-reticulares, y de las fibras profundas del fascículo longitudinal superior. En el cuerpo calloso se objetiva un descenso en el número de fibras comisurales, que son reemplazadas por gliosis. El FSC se encuentra alterado, con un patrón de última pradera en la región subcortical adyacente a los ventrículos, correspondiente a la intersección entre las arterias subependimarias y las arterias perforantes dependientes de los grandes troncos arteriales de la circulación anterior. El recambio diario del LCR se ve disminuido en un 75%, lo que conlleva una reducción del aclaramiento de neurotóxicos y la interrupción de las señalizaciones neuroendocrinas y paracrinas que ocurren a través del LCR.
ConclusionesLa HCAi emerge como una entidad nosológica compleja, en la que los efectos de la microangiopatía subcortical y la disminución del recambio de LCR desempeñan un papel fundamental. Esta base fisiopatológica aleja la HCAi del concepto clásico de hidrocefalia y la acerca al perfil de otras enfermedades neurodegenerativas, como la enfermedad de Alzheimer o la enfermedad de Binswanger.
Since its description five decades ago, the pathophysiology of idiopathic chronic adult hydrocephalus (iCAH) has been traditionally related to the effect that ventricular dilatation exerts on the structures surrounding the ventricular system. However, altered cerebral blood flow, especially a reduction in the CSF turnover rate, are starting to be considered the main pathophysiological elements of this disease.
DevelopmentCompression of the pyramidal tract, the frontostriatal and frontoreticular circuits, and the paraventricular fibres of the superior longitudinal fasciculus have all been reported in iCAH. At the level of the corpus callosum, gliosis replaces a number of commissural tracts. Cerebral blood flow is also altered, showing a periventricular watershed region limited by the subependymal arteries and the perforating branches of the major arteries of the anterior cerebral circulation. The CSF turnover rate is decreased by 75%, leading to the reduced clearance of neurotoxins and the interruption of neuroendocrine and paracrine signalling in the CSF.
ConclusionsiCAH presents as a complex nosological entity, in which the effects of subcortical microangiopathy and reduced CSF turnover play a key role. According to its pathophysiology, it is simpler to think of iCAH more as a neurodegenerative disease, such as Alzheimer disease or Binswanger disease than as the classical concept of hydrocephalus
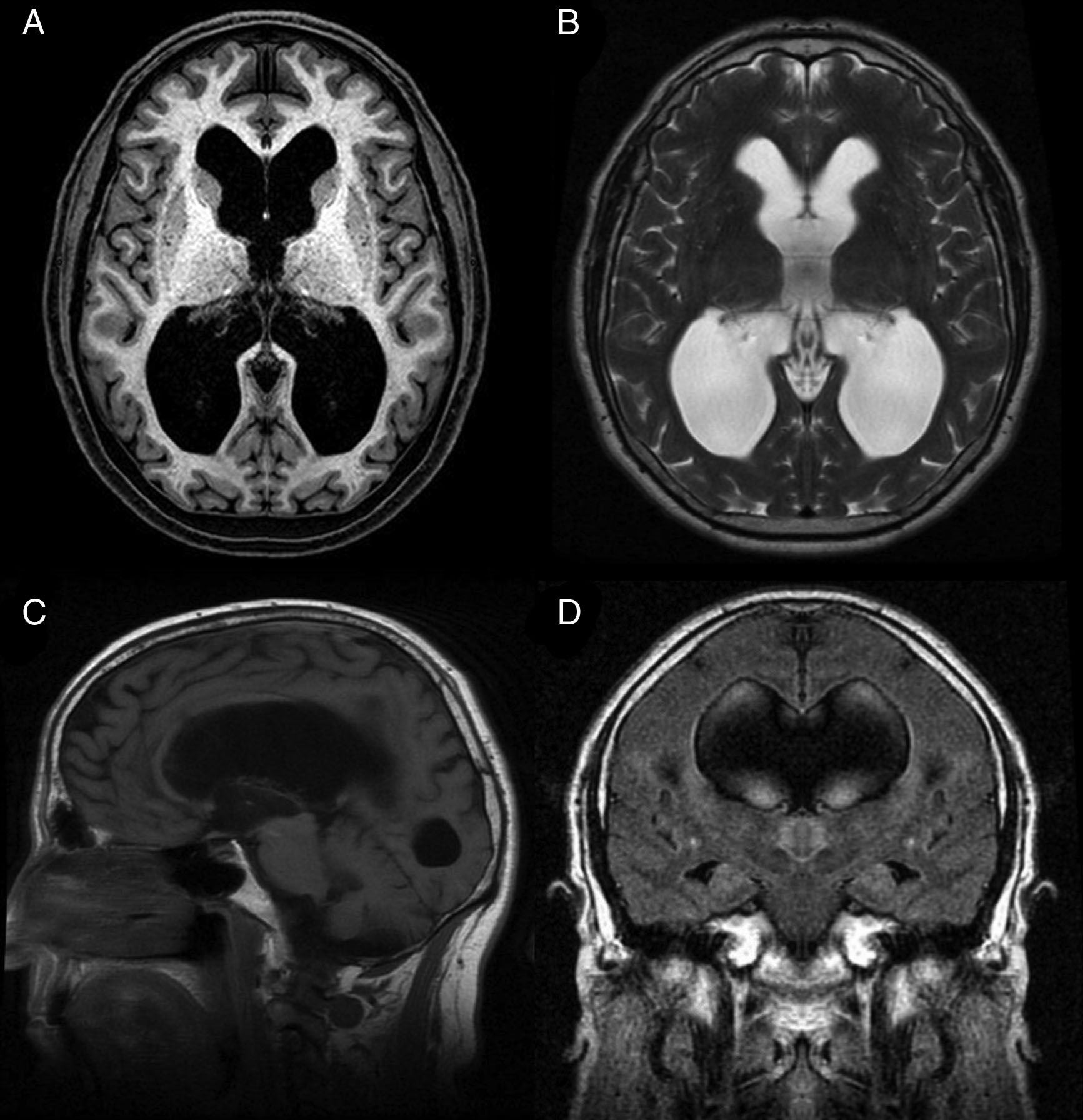
La hidrocefalia crónica del adulto idiopática (HCAi) es una entidad nosológica caracterizada por la tríada clínica de trastorno de la marcha, deterioro cognitivo e incontinencia urinaria, en presencia de dilatación del sistema ventricular en las pruebas de neuroimagen (fig. 1) y en ausencia de otra causa que justifique los hallazgos clínicos.
 Corte axial potenciado en T1. Moderada dilatación de los ventrículos laterales y del tercer ventrículo. B) Corte axial potenciado en T2. Artefactos de señal por vacío de flujo en el tercer ventrículo y ausencia de hiperintensidades de señal tanto a nivel periventricular como subcortical. C) Corte sagital potenciado en T1. Descenso del suelo y abalonamiento del tercer ventrículo, con reducción de la distancia mamilo-pontina. El acueducto de Silvio se encuentra permeable, sin que se visualicen causas obstructivas que justifiquen la dilatación ventricular. D) Corte coronal FLAIR. Patrón típico de borramiento de surcos en la convexidad alta del cerebro, especialmente en la línea media. El artefacto por vacío de flujo que se objetivaba en la secuencia T2 se extiende hacia ambos agujeros de Monro, alcanzando la porción inferior y medial de los cuerpos ventriculares. Ausencia de hiperintensidades de señal periventriculares y subcorticales.")
Resonancia magnética preoperatoria de un paciente diagnosticado de hidrocefalia crónica del adulto idiopática con buena respuesta tras la implantación de una derivación ventrículo-peritoneal. A) Corte axial potenciado en T1. Moderada dilatación de los ventrículos laterales y del tercer ventrículo. B) Corte axial potenciado en T2. Artefactos de señal por vacío de flujo en el tercer ventrículo y ausencia de hiperintensidades de señal tanto a nivel periventricular como subcortical. C) Corte sagital potenciado en T1. Descenso del suelo y abalonamiento del tercer ventrículo, con reducción de la distancia mamilo-pontina. El acueducto de Silvio se encuentra permeable, sin que se visualicen causas obstructivas que justifiquen la dilatación ventricular. D) Corte coronal FLAIR. Patrón típico de borramiento de surcos en la convexidad alta del cerebro, especialmente en la línea media. El artefacto por vacío de flujo que se objetivaba en la secuencia T2 se extiende hacia ambos agujeros de Monro, alcanzando la porción inferior y medial de los cuerpos ventriculares. Ausencia de hiperintensidades de señal periventriculares y subcorticales.
Aunque parecen existir descripciones previas de este síndrome clínico1-5 —entre las que caben destacar la realizada por el neurólogo francés Etienne Mouline6 en 1819 o la efectuada por el patólogo alemán Friedrich Dörner7 en 1826—, fue el recientemente fallecido neurocirujano colombiano Salomón Hakim Dow el que, hace más de 5 décadas, sistematizara en su tesis doctoral los hallazgos clínicos y radiológicos que lo conforman8, y lo diese a conocer, en colaboración con los conocidos neurólogos del Massachusetts General Hospital Raymond D. Adams y Charles M. Fisher, en 2 artículos publicados de forma simultánea en el New England Journal of Medicine9 y el Journal of Neurological Sciencies10.
Tradicionalmente, siempre se ha intentado explicar el mecanismo por el que se producen los 3 ejes sintomáticos de la HCAi acudiendo al efecto que la propia dilatación ventricular ejerce sobre las estructuras nerviosas11-17 y vasculares periventriculares18-25. Sin embargo, estudios recientes parecen implicar también la incapacidad del LCR para drenar las sustancias de desecho del líquido intersticial en el desarrollo de la clínica de esta enfermedad26-29.
En este artículo realizamos una actualización de la fisiopatología de esta enfermedad, poniendo especial énfasis en la reducción del recambio licuoral como un aspecto novedoso que podría condicionar la evolución a largo plazo de la HCAi, acercando más esta entidad al perfil de una enfermedad neurodegenerativa que al concepto clásico de hidrocefalia que manejamos habitualmente.
DesarrolloCompresión de las fibras subcorticales periventricularesLa compresión de las proyecciones frontales que descienden en proximidad a las astas frontales de los ventrículos laterales provoca su alteración funcional, que puede ser completamente reversible si la disfunción se debe al enlentecimiento o detención del flujo axoplásmico a dicho nivel, o permanente cuando se produce desmielinización o perdida de la integridad axonal16,17. Las diferentes técnicas de resonancia magnética han permitido profundizar en el conocimiento de los fascículos afectados, incluyendo las proyecciones corticales y subcorticales de estos, y determinar si su lesión es reversible o irreversible. Para ello se utilizan básicamente las secuencias ponderadas en difusión (diffusion weighted imaging) y las secuencias de tensor de difusión (diffusion tensor imaging [DTI]). Con la primera de ellas es posible valorar la presencia de agua libre a nivel intersticial mediante los coeficientes de difusión aparente (apparent diffusion coefficients [ADC]), ya que se elevan en el edema extracelular. Sin embargo, la sustitución de axones por gliosis —que invariablemente ocurre en la fase crónica del daño axonal— también puede provocar un aumento del ADC, inclusive de mayor calibre. Las secuencias DTI, incluida la tractografía de fibras, son mucho más sensibles que los coeficientes de difusión a la hora de evaluar la integridad de los fascículos nerviosos, su densidad y su posible desplazamiento, al detectar los cambios en la anisotropía del movimiento de las moléculas de agua debida a la unidireccionalidad de los axones. La difusibilidad media (mean diffusivity [MD]) puede interpretarse de forma análoga a los ADC, mientras que la anisotropía fraccional (fractional anisotropy [FA]) disminuye significativamente cuando se produce una disrupción axonal, inclusive en zonas en las que los mapas ADC no muestran una gran elevación de los coeficientes de difusión o estos son normales debido al artefacto generado por la gliosis residual, y aumenta cuando la densidad de los axones se incrementa, por ejemplo, como consecuencia de una compactación de los mismos por efecto de un vector de carga perpendicular a su trayectoria. Consecuentemente, la presencia de un incremento en el ADC o en la MD, manteniendo valores normales o aumentados de la FA resulta indicativa de edema intersticial, mientras que la disminución de la FA debería ser interpretada como lesión axonal con independencia de los valores de MD o ADC30.
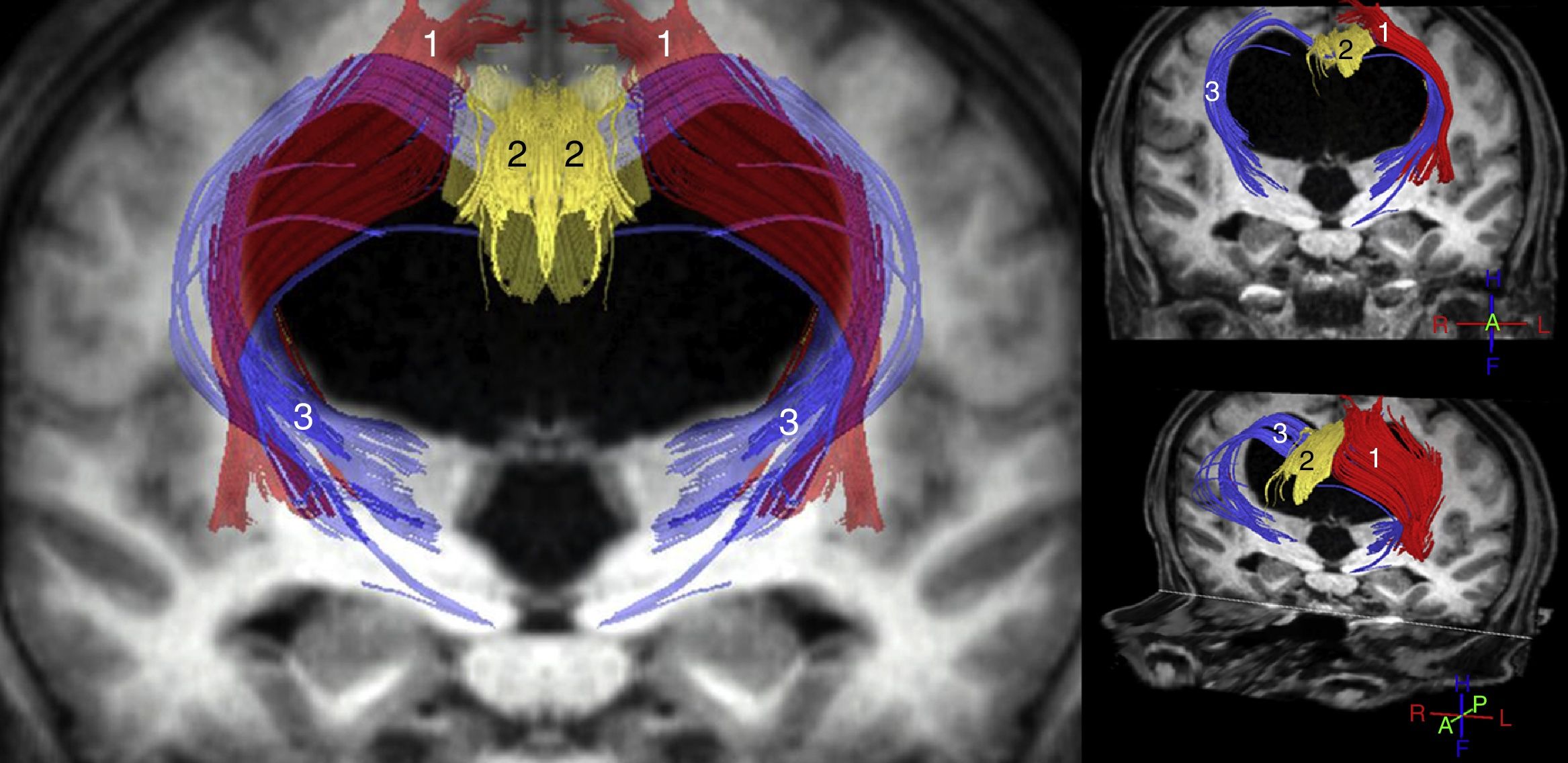
Los estudios más importantes realizados hasta el momento con dichas técnicas parecen coincidir en que el ADC y/o el MD aumentan tanto en el cuerpo calloso como en la cápsula blanca interna. La FA, sin embargo, se encuentra incrementada en la cápsula blanca interna, especialmente en su brazo anterior, y disminuida en el cuerpo calloso, sobre todo a nivel de la rodilla del mismo11-14,31. Hallazgos adicionales han sido publicados por Osuka et al.11,31, que mostraron una elevación de la FA a nivel del núcleo caudado; por Lenfeldt et al.,15 que objetivaron un aumento del MD sin cambios en la FA a nivel de la sustancia blanca asociada a la corteza precentral, y por Kamiya et al., que demostraron un aumento de la densidad axonal del fascículo cortico-espinal a nivel paraventricular, consecuencia de su compactación en el trayecto adyacente al ventrículo32. A la luz de estos datos, se puede concluir que en la HCAi existe una compresión del tracto piramidal y de los circuitos córtico-subcórtico-corticales frontoestriatales y fronto-reticulares (fig. 2), así como una afectación de las fibras profundas del fascículo longitudinal superior. En el cuerpo calloso la disminución de la FA y el incremento del ADC y/o MD resultan compatibles con un descenso en el número de fibras comisurales, que habrían sido reemplazadas por gliosis.
 del paciente mostrado en la figura 1, en la que puede observarse la distorsión generada por la dilatación ventricular en el trayecto del tracto piramidal (1) y en las conexiones córtico-estriadas y córtico-reticulares (3). Estos últimos tractos se encuentran adelgazados de una forma bastante acusada, al igual que ocurre con el fascículo cingulado (2) y, en menor medida, con el tracto piramidal (1).")
Reconstrucción tractográfica de las secuencias de tensor de difusión (DTI) del paciente mostrado en la figura 1, en la que puede observarse la distorsión generada por la dilatación ventricular en el trayecto del tracto piramidal (1) y en las conexiones córtico-estriadas y córtico-reticulares (3). Estos últimos tractos se encuentran adelgazados de una forma bastante acusada, al igual que ocurre con el fascículo cingulado (2) y, en menor medida, con el tracto piramidal (1).
Una gran variedad de estudios han demostrado que el FSC se encuentra disminuido en la HCAi, aunque la afectación global es más bien discreta, con una reducción del 20-30%18-25,33. En un reciente trabajo de la Universidad de Osaka ha podido objetivarse que el FSC es comparativamente bajo con respecto a los controles normales, no solo en aquellos pacientes con una HCAi confirmada, sino también en individuos con una dilatación ventricular con características radiológicas de HCAi pero que aún no han desarrollado el síndrome clínico25. Con respecto a la intensidad de la clínica y su relación con el FSC, los resultados son contradictorios, ya que la mayor parte de los estudios no demuestran una dependencia directa18,22,25, mientras que otros correlacionan la gravedad de la clínica con el descenso progresivo del FSC19,21,34.
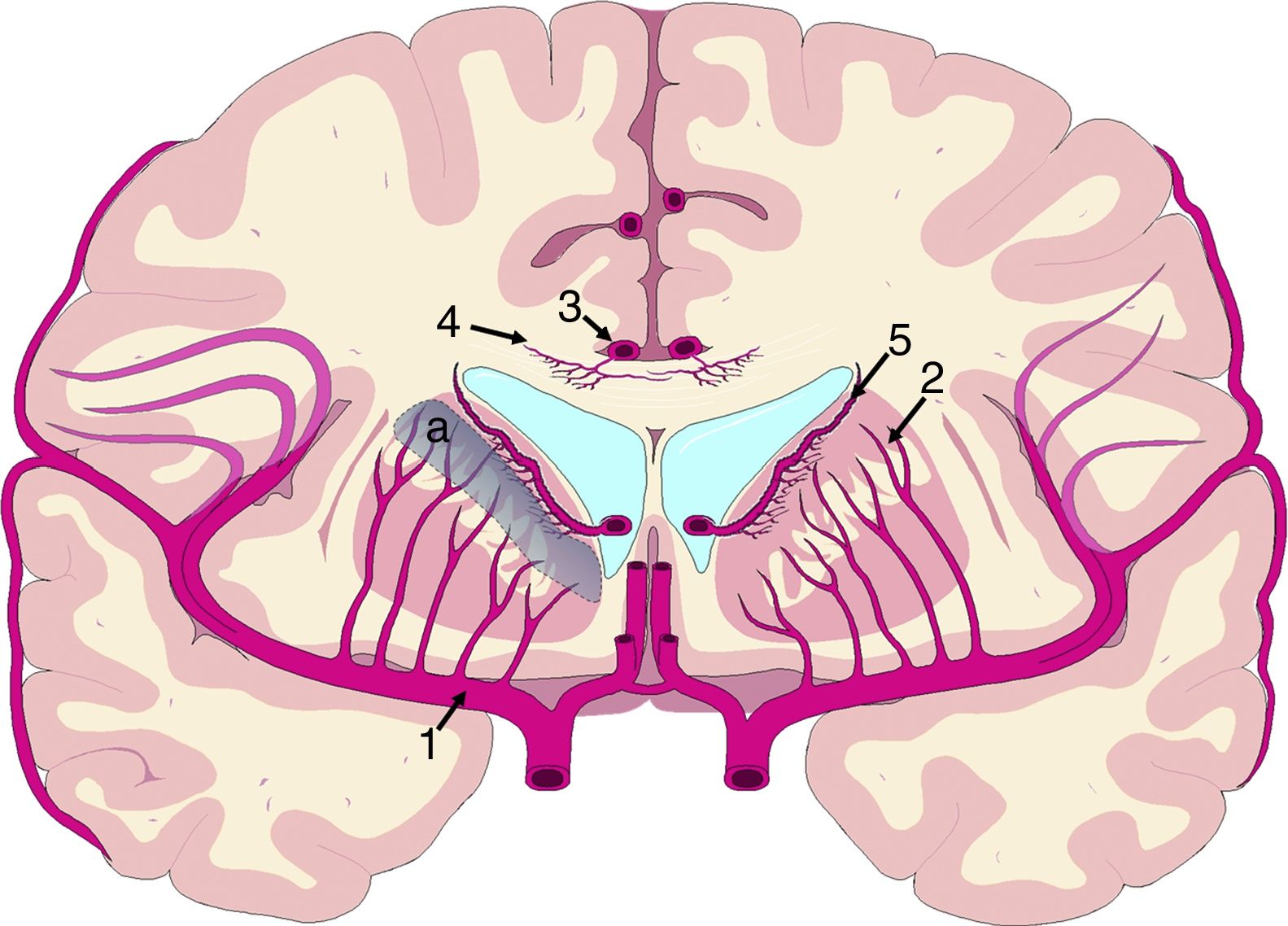
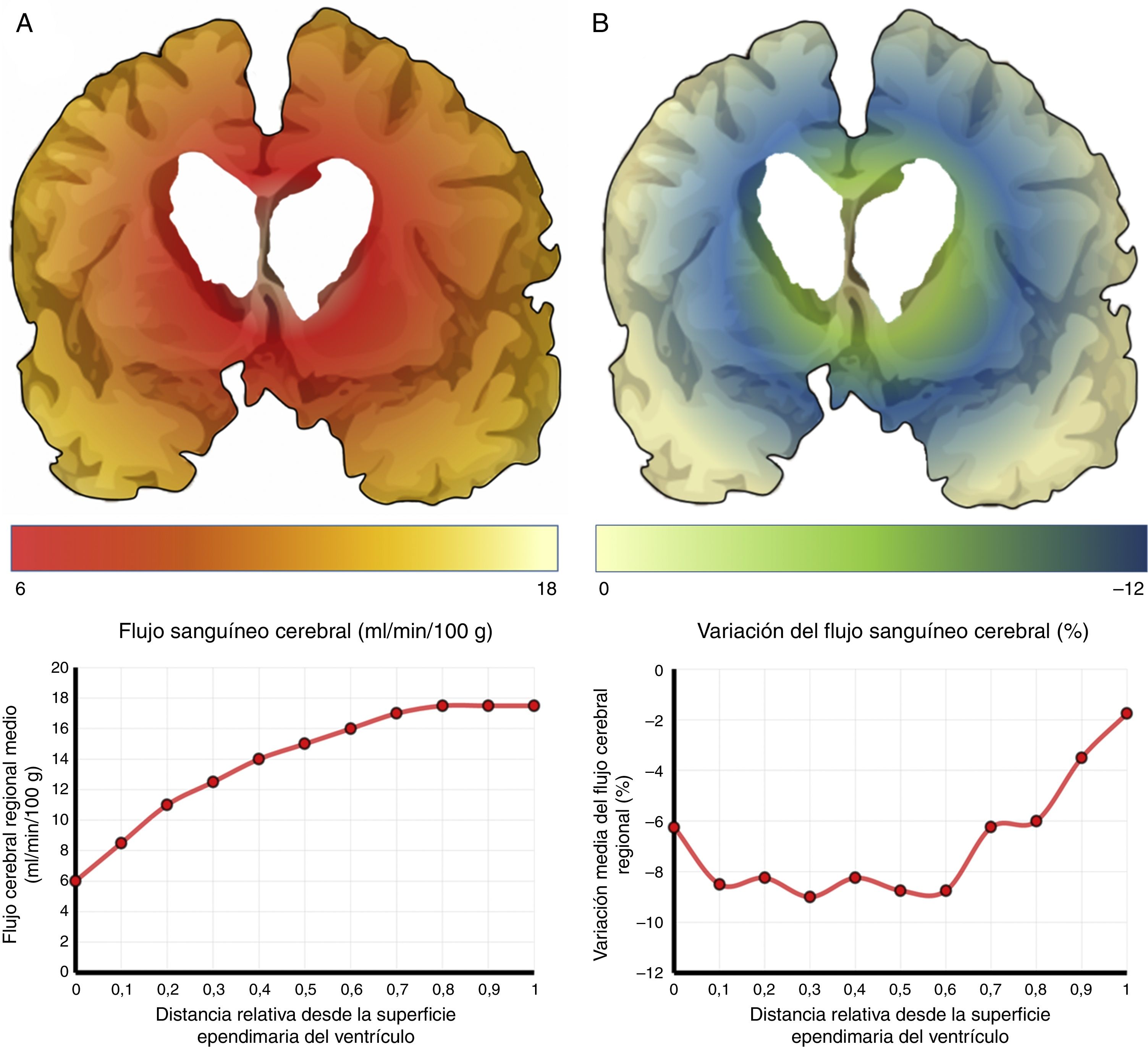
El mecanismo de la alteración del FSC no se ha dilucidado completamente. Si la disminución del FSC fuese debida únicamente a la distorsión que la dilatación ventricular y los vectores de carga ejercen sobre la microcirculación cerebral, cabría esperar que se relacionase de forma inversa con la distancia al sistema ventricular, existiendo mayor caída del FSC en la porción más interna e inferior del centro semioval, en el tálamo y en la cabeza y la cola del núcleo caudado. Algunos estudios recientes, que han logrado superar las limitaciones de resolución espacial de las técnicas clásicas para la medición del FSC empleando corregistro de resonancia magnética con tomografía por emisión de positrones con agua marcada con 15O, parecen demostrar la existencia de un gradiente de FSC desde la región periventricular hasta el córtex, aunque la relación no es proporcional a la distancia18,19. El trabajo de Momjian et al. resulta particularmente interesante en este sentido, ya que localiza la región de máxima afectación de la sustancia blanca subcortical a un cm de la pared ventricular, determinando además una disminución de la reserva cerebrovascular (RCV) a dicho nivel de más del 50% (fig. 3). Sus resultados son compatibles con un fenómeno de última pradera en la región subcortical adyacente a los ventrículos, fenómeno que cuenta con una base microanatómica coherente, ya que el tejido más cercano al epéndimo se encuentra irrigado por las arterias subependimarias y el más distal por arterias perforantes dependientes de las grandes troncos arteriales de la circulación anterior (fig. 4)35,36. La zona de máxima sensibilidad a la isquemia es precisamente el área de intersección entre ambas regiones de vascularización y es donde se produce la máxima alteración tanto del FSC como de la RCV. De todas formas, la afectación de los núcleos de la base parece repetirse en la mayor parte de los estudios clínicos de HCAi19,34,37.
 entre el territorio de las ramas perforantes (2) de la arteria cerebral media (1) y las ramificaciones de las arterias subependimarias (5). El cuerpo calloso se encuentra irrigado fundamentalmente por arterias callosas cortas (4), dependientes de la arteria pericallosa (3), con carácter de circulación terminal.")
Vascularización de las estructuras periventriculares. Los núcleos de la base y la cápsula blanca interna se encuentran en una región de última pradera (a) entre el territorio de las ramas perforantes (2) de la arteria cerebral media (1) y las ramificaciones de las arterias subependimarias (5). El cuerpo calloso se encuentra irrigado fundamentalmente por arterias callosas cortas (4), dependientes de la arteria pericallosa (3), con carácter de circulación terminal.
 de la región periventricular en la hidrocefalia crónica del adulto idiopática. A) Relación entre el FSC y la distancia desde la superficie ependimaria del ventrículo, mostrando una curva de tipo logaritmo18. B) Cuando se produce una sobrecarga volumétrica, el FSC cae en la zona de última pradera subependimaria, situada entre 5 y 15mm de la superficie ventricular.")
Flujo sanguíneo cerebral (FSC) de la región periventricular en la hidrocefalia crónica del adulto idiopática. A) Relación entre el FSC y la distancia desde la superficie ependimaria del ventrículo, mostrando una curva de tipo logaritmo18. B) Cuando se produce una sobrecarga volumétrica, el FSC cae en la zona de última pradera subependimaria, situada entre 5 y 15mm de la superficie ventricular.
Sin embargo, también se han detectado alteraciones en áreas corticales que no pueden justificarse por un simple fenómeno mecánico, dada su relativa lejanía del sistema ventricular. Las regiones mesiales anterior e inferior del lóbulo frontal presentan una hipoperfusión más acusada que el resto de las estructuras cerebrales22,34, habiéndose descrito alteraciones en otras áreas, como el córtex anterior temporal izquierdo22, la región hipocampal y parahipocampal37, la sustancia blanca del lóbulo frontal correspondiente al fascículo longitudinal superior34 o las áreas asociativas parietales34.
La afectación cerebrovascular en la HCAi es más amplia e implica también la vasorreactividad cerebral, que se encuentra disminuida como manifestación de una RCV extenuada. Chang et al. realizaron un estudio de FSC mediante SPECT con 99mTc-HM-PAO en 167 pacientes con diagnóstico de HCAi, antes y después de la administración de 1g de acetazolamida, con el fin de valorar la RCV. Demostraron que el incremento de FSC tras la infusión de acetazolamida era un 50-80% menor que el que ocurría en sujetos normales, pudiendo corresponderse los resultados a una disminución de la vasorreactividad frente al aumento de la PaCO2 o a una RCV significativamente reducida21. Afortunadamente estos cambios no se acompañan de una alteración en el acoplamiento metabólico de las áreas implicadas, ya que la tasa metabólica cerebral se encuentra reducida de una forma proporcional a la reducción del FSC y la fracción de extracción de oxígeno permanece dentro de límites normales38,39.
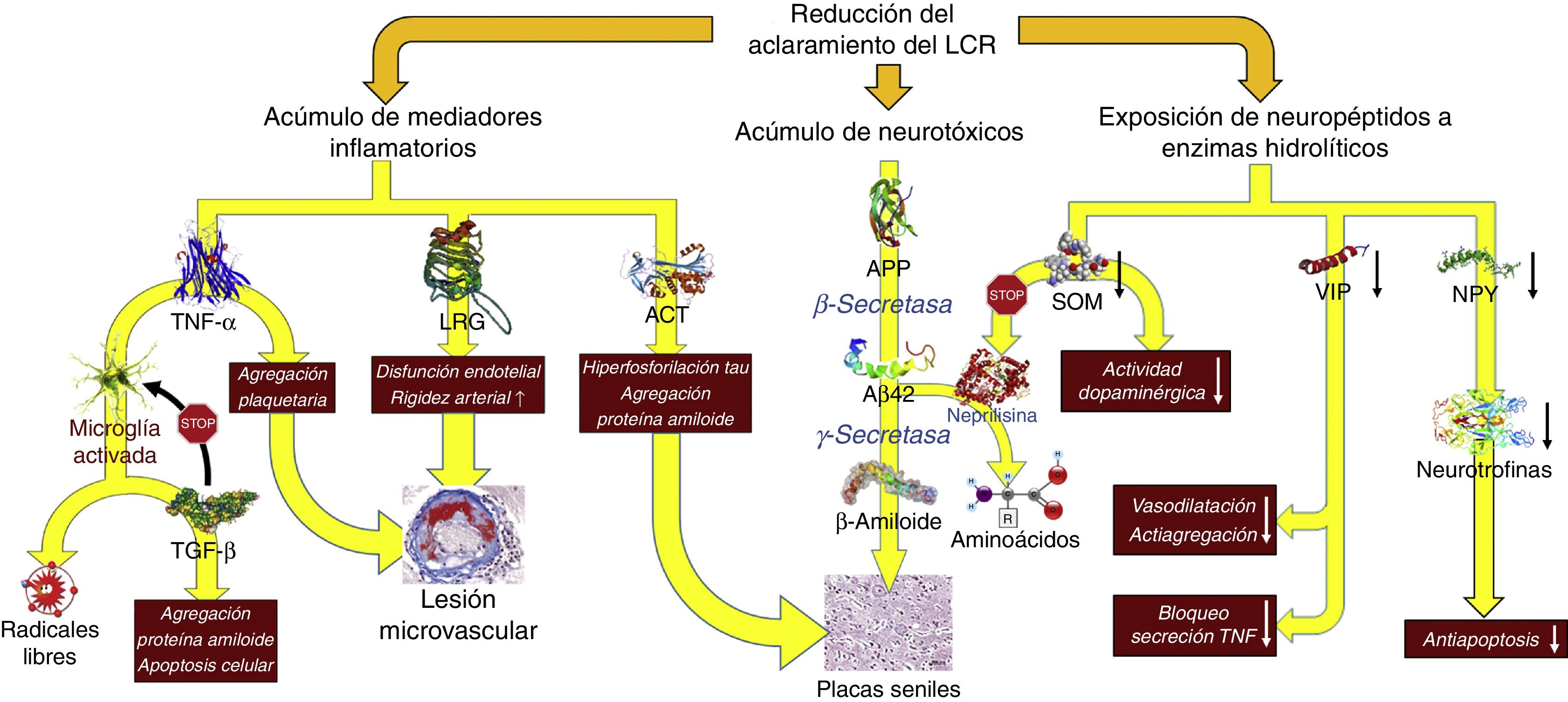
Reducción del recambio licuoral (fig. 5)De forma similar al comportamiento de los linfáticos en el resto del organismo, el LCR es el encargado de la excreción de las macromoléculas presentes en el fluido intersticial que no pueden reabsorberse en el polo venoso del capilar. Para entender el funcionamiento de este sistema de excreción debemos de tener en cuenta que, en condiciones normales, las más de 2.000 proteínas diferentes que se encuentran en el LCR representan menos del 4‰ de su peso40, y que por tanto la reabsorción del agua del LCR puede estar desligada de la excreción de macromoléculas: la primera puede realizarse perfectamente en el polo venoso del capilar, mientras que la segunda tendrá lugar en las granulaciones de Pacchioni o en los linfáticos basicraneales. La alteración persistente de la excreción de macromoléculas es posible aun cuando se llegue a un equilibrio volumétrico en la producción-reabsorción del componente acuoso del LCR.

Consecuencias fisiopatológicas de la reducción del aclaramiento licuoral, con acúmulo de mediadores inflamatorios y neurotoxinas, y exposición de los neuropéptidos responsables de la señalización neuroendocrina a la actividad enzimática hidrolítica.
Aβ42: fragmento de 42 residuos de la proteína β amiloide; ACT: antiquimotripsina; APP: proteína precursora del amiloide; LRG: glicoproteína rica en leucina; NPY: neuropéptido Y; TGF: factor de crecimiento transformante; TNF: factor de necrosis tumoral. VIP: péptido intestinal vasoactivo.
En la HCAi la tasa de producción de LCR se encuentra disminuida hasta alcanzar los 0,25±0,08ml/min, es decir, más de un 30%41. Adicionalmente, el aumento del tamaño ventricular hace que el volumen de distribución se incremente un 30%, sobrepasando los 200ml en la mayor parte de los pacientes. Estas circunstancias, asociadas a la presencia de un trastorno de la reabsorción licuoral, hacen que el recambio diario del LCR se vea reducido en un 75%. Sin embargo, los cambios adaptativos que acontecen tanto en el plexo coroideo como en los pies vasculares de los astrocitos impiden que el tándem producción-reabsorción acuosa se desequilibre. Por un lado, disminuye la expresión de la isoforma 1 de la acuaporina en el plexo coroideo42-44 mediante la activación del sistema de péptidos natriuréticos de los órganos circunventriculares y de los núcleos hipotalámicos45,46, lo que se traduce en una reducción del transporte de agua en la membrana apical del epitelio coroideo y, en consecuencia, en una disminución en la producción de LCR. Por otro lado, la expresión de la isoforma 4 de la acuaporina en los astrocitos de la sustancia blanca se ve incrementada42,43,47-49, probablemente en un intento de aumentar la reabsorción del componente acuoso del LCR en el polo venoso capilar42,50. No obstante, este incremento de absorción a nivel del polo venoso puede reducir la presión intersticial en la región periventricular, favoreciendo la aparición de un gradiente de presión entre el intersticio y el ventrículo, y perpetuando la dilatación ventricular51,52. Por el momento, no existen datos que permitan sacar conclusiones de cómo esta situación afecta al aclaramiento de proteínas en la HCAi, aunque es plausible que la alteración del recambio licuoral tenga al menos 2 vertientes que deban ser consideradas: la reducción del aclaramiento de neurotóxicos y la interrupción de las señalizaciones neuroendocrinas y paracrinas que ocurren a través del LCR53.
No existe evidencia directa de una reducción del aclaramiento de neurotóxicos en la HCAi, aunque es posible obtenerla indirectamente de los estudios con biomarcadores y del análisis proteómico del LCR. La mayor parte de los autores demuestran una disminución de la mayor parte de los metabolitos de la vía de procesamiento proteolítico del amiloide, incluida la proteína precursora del β-amiloide (APP) y del péptido Aβ4254-58. La proteína Tau y su variante fosforilada se mantienen dentro de rangos normales o inclusive disminuidos54,57,59. La reducción del recambio licuoral provocaría un déficit en el aclaramiento intersticial de APP, permitiendo su procesamiento secuencial primero por la β-secretasa y después por la γ-secretasa para dar lugar a Aβ42, que se depositaría en forma de placas de amiloide, disminuyendo la concentración licuoral de estos 2 compuestos26-29. El estudio realizado por Fagan et al.60 en sujetos sin demencia ha dado cuerpo a estas especulaciones, demostrando que aquellos individuos con depósitos de amiloide cerebral, evidenciados mediante PET con PiB (Pittsburg compund B) marcado con 11C, tenían niveles más bajos de Aβ42 en LCR. Pyykoö et al.61 confirman estos hallazgos en pacientes afectados de HCAi, objetivando una relación inversamente lineal entre la carga de proteína amiloide en la biopsia cerebral y la concentración de Aβ42, tanto en el LCR ventricular como en el extraído de la teca lumbar. Adicionalmente, Moriya et al.62 y Jeppsson et al.58 han observado un aumento de los niveles de Aβ42 en el LCR de pacientes diagnosticados de HCAi tras la implantación de un sistema derivativo, correlacionándose su incremento con el grado de mejoría neurológica que experimentaron los enfermos. Estos hallazgos parecen confirmar que la normalización de la dinámica licuoral tras la cirugía de derivación promueve el desplazamiento de la proteína β-amiloide de su forma oligomérica a su forma monomérica como consecuencia del incremento en el isómero Aβ38, de baja agregabilidad62.
Datos adicionales aparecen de mano del perfil inflamatorio licuoral, ya que la presencia de un exceso de reactantes de fase aguda en el LCR —en ausencia de una celularidad anormal en el análisis bioquímico o de activación microglial en la región periventricular que hagan sospechar un proceso inflamatorio como etiología de los hallazgos— sugiere una afectación del aclaramiento licuoral como responsable del acúmulo de mediadores proinflamatorios de origen astrocitario. Así se ha demostrado un aumento de α1-glicoproteína rica en leucina (leucine rich α2-glycoprotein)63,64, α1-antiquimotripsina63,65, haptoglobina63, isoforma 1 de la transferrina66, α1β1 glicoproteina65 o factor de necrosis tumoral α (tumoral necrosis factor)67. También se ha observado un incremento en los productos de la acción de los radicales libres, dato que igualmente podría considerarse a favor de una disminución del recambio licuoral68.
Li et al. describieron también un incremento de la señalización dependiente de factor de crecimiento transformante β (transforming growth factor [TGF-β])69. Aunque es posible que un porcentaje del exceso de TGF-β se deba también a un déficit en su aclaramiento, este no explicaría la regulación al alza del subtipo 2 de su receptor que también se objetivó en el mencionado trabajo, por lo que la presencia de un mecanismo adaptativo en respuesta a la presencia creciente de mediadores y reactantes inflamatorios es también posible, ya que esta citocina ejerce un efecto protector bloqueando la respuesta inflamatoria neuroglial y endotelial70-72.
Los efectos potenciales del anormal acúmulo de estas proteínas resultan evidentes:
- –
La disminución del aclaramiento de APP favorecería el depósito vascular y tisular de amiloide, aumentando la velocidad de desarrollo de otros procesos neurodegenerativos intercurrentes o propiciando su aparición (p. ej., enfermedad de Alzheimer)53.
- –
La difusión de proteínas proinflamatorias en la región periventricular, especialmente de TNF-α —que actúa como proagregante y favorece la adhesión celular en los capilares— alteraría la dinámica microvascular, acentuando el efecto mecánico de la dilatación ventricular en la región periventricular18.
- –
Los radicales libres resultan lesivos para neuronas, glía y endotelio, y su efecto se ha relacionado a nivel molecular con la fisiopatología de una gran variedad de enfermedades neurológicas, resultando más que plausible su implicación en el daño tisular de la HCAi68.
- –
El TGF-β promueve la apoptosis tanto de neuronas como de oligodendrocitos73,74. La muerte celular programada diferida podría explicar el progresivo declinar clínico que en no pocas ocasiones se objetiva en estos pacientes, aun a pesar de una derivación licuoral inicialmente exitosa y correctamente funcionante.
La alteración de la señalización neuroendocrina licuoral también es un mecanismo a considerar, a pesar de la ausencia de pruebas objetivas que demuestren su existencia de forma fehaciente. Se han descrito más de 100 neuropéptidos que podrían utilizar la circulación del LCR para llegar a regiones distantes del sistema nervioso central y que presentan unas características propias en cuanto a su síntesis, liberación y regulación, jugando además funciones radicalmente diferentes con respecto a los neurotransmisores convencionales75.
Se han constatado niveles bajos en el LCR de somatostatina (SOM)76-80, péptido intestinal vasoactivo (VIP)59,79,81, neuropéptido Y (NPY)59,79,80,82, colecistoquinina83, péptido inductor del sueño δ79,84 y hormona liberadora de corticotropina80. La interpretación de estos hallazgos es compleja, ya que pueden ser la consecuencia de una disfunción prácticamente global de los sistemas de neurotransmisión peptidérgica secundaria a la propia enfermedad, o reflejar un mecanismo activo en el que el efecto del deficiente recambio licuoral dejaría expuestos estos péptidos a la acción de las neuropeptidasas, disminuyendo por tanto sus niveles en el LCR e interrumpiendo la neurotransmisión por vía licuoral85,86.
Probablemente el péptido mejor estudiado en este sentido sea la SOM, que se localiza de forma prácticamente difusa en todo el cerebro, aunque su producción principal radica en los núcleos preóptico, paraventricular, arcuato y ventromedial del hipotálamo. Además de su acción reguladora endocrina, como péptido antagonista de la hormona liberadora de somatotropina, la SOM facilita la transmisión dopaminérgica en el estriado y contribuye al normal funcionamiento de las neuronas durante el envejecimiento87. Esta última función se encuentra ligada a la actividad proteolítica de la neprilisina, una de las proteasas encargadas del catabolismo de la Aβ42 y cuya actividad induce la SOM88. La disminución de la actividad de la SOM secundaria a un deficiente recambio licuoral podría favorecer el acúmulo de Aβ42 y empeorar el cuadro clínico. El NPY también se distribuye de forma difusa en el encéfalo, incluyendo el complejo amigdalohipocampal, los núcleos de la base y, por supuesto, el hipotálamo, donde se colocaliza con las neuronas productoras de SOM en los núcleos paraventricular y arcuato87. Un reciente artículo ha demostrado que el NPY equilibra los efectos tóxicos de la Aβ promoviendo la síntesis celular de neurotrofinas89, por lo que su disminución en la HCAi favorecería la pérdida de elementos neurales, máxime si tenemos en consideración el efecto sinérgico de la SOM anteriormente descrito. Un efecto similar ejerce el VIP que, además de su función vasodilatadora y de inducir la síntesis glial de neurotrofina-3 y del factor neurotrófico dependiente de actividad (activity-dependent neurotrophic factor [ADNF]), inhibe la respuesta inflamatoria neuroglial, sobre todo al bloquear la producción microglial de TNF-α y radicales libres90.
ConclusionesLa fisiopatología de la HCAi no solamente descansa sobre la compresión de las fibras subcorticales periventriculares. Las alteraciones del FSC en las áreas de última pradera entre las arterias perforantes dependientes de los grandes troncos arteriales de la circulación anterior y las arterias subependimarias, así como la reducción del recambio licuoral, que conlleva una disminución en el aclaramiento de neurotóxicos y la interrupción en la señalización neuroendocrina licuoral, emergen como piezas claves que permiten explicar la peculiar evolución clínica a largo plazo que presenta este tipo de hidrocefalia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

