





La APP es un síndrome clínico caracterizado por un deterioro progresivo del lenguaje de etiología neurodegenerativa. En los últimos años se han realizado importantes avances que han contribuido a un mayor conocimiento de esta entidad, que puede ser el modo de presentación de diferentes enfermedades neurodegenerativas.
DesarrolloSe revisan los principales aspectos epidemiológicos, clínicos, diagnósticos, etiológicos y terapéuticos de la APP. La mayoría de los casos son esporádicos, iniciándose en torno a los 50-70 años. Se han descrito 3 subtipos clínicos principales: APP-no fluente o agramatical; APP-semántica; y APP-logopénica. Cada subtipo se ha asociado de forma preferencial a una anatomía patológica concreta, aunque la capacidad predictiva del diagnóstico clínico no es completa. Entre los biomarcadores disponibles, destacan la neuroimagen anatómica y funcional. Se han ensayado diferentes tratamientos, sin demostrarse un beneficio claro. No obstante, los inhibidores de acetilcolinesterásica pueden estar indicados, especialmente en la variante logopénica.
ConclusionesLa APP es un síndrome emergente, con una prevalencia probablemente mayor de la esperada. Considerada previamente como parte del espectro de la demencia frontotemporal, también está relacionada con la enfermedad de Alzheimer. El diagnóstico clínico, completado con el uso de biomarcadores, puede predecir la anatomía patológica subyacente, lo que a su vez supondrá mayores oportunidades en el tratamiento.
Primary progressive aphasia (PPA) is a clinical syndrome characterised by a progressive decline in language and speech of neurodegenerative origin. Major breakthroughs made in recent years have lent us a better understanding of this syndrome, which may be the first manifestation of any of a number of neurodegenerative diseases.
DevelopmentWe reviewed the main aspects of PPA epidemiology, clinical manifestations, diagnosis, aetiology and treatment. Most cases manifest sporadically and the typical age of onset is between 50 and 70 years. Three clinically distinct variants have been described: nonfluent or agrammatic PPA, semantic PPA and logopenic PPA. Each of these variants tends to be associated with specific histopathological findings, but clinical diagnostic methods are imperfect predictors of underlying pathology. Anatomical and functional neuroimaging can provide useful biomarkers. Several treatments have been proposed, and while no clear benefits have been demonstrated, acetylcholinesterase inhibitors may be useful, especially in the logopenic variant.
ConclusionsPPA is an emerging syndrome which may be more prevalent than we might expect. It was previously listed as part of the frontotemporal dementia spectrum, and it is also related to Alzheimer disease. Clinical diagnosis, complemented by a biomarker evaluation, may predict the underlying pathology, which in turn will improve treatment possibilities.
La afasia progresiva primaria (APP) es un síndrome clínico caracterizado por un deterioro insidioso del lenguaje de etiología neurodegenerativa. La alteración del lenguaje debe ser la principal manifestación clínica durante al menos los 2 primeros años de evolución, estando el resto de dominios cognitivos (memoria, habilidades visuoespaciales, etc.) preservados, y justificándose la limitación en las actividades de la vida diaria únicamente por la disfunción del lenguaje. Descrito en la literatura médica reciente por Mesulam, se trata de síndrome heterogéneo que puede ser el modo de presentación de diversas enfermedades neurodegenerativas. En los últimos años se ha profundizado, especialmente, en la clasificación de la APP en varias formas clínicas y la descripción de biomarcadores, lo que puede contribuir a predecir la anatomía patológica. Con este artículo pretendemos revisar los principales avances realizados en el conocimiento de la APP en los últimos años, avances que han modificado sustancialmente su abordaje clínico y plantean interesantes perspectivas para el futuro.
Epidemiología y factores de riesgoLa APP suele empezar entre los 50 y los 70 años1. No existen claras diferencias entre varones y mujeres, siendo ligeramente más frecuente en varones2 o mujeres3 según los estudios. La frecuencia en la población general es desconocida, extrapolándose a partir de los datos disponibles para el grupo de demencia frontotemporal. A partir de esto, la prevalencia de DFT se calcula en aproximadamente 5 casos por millón de habitantes4, entre 1-15 casos/100.000 habitantes en el grupo de población menor de 65 años5–7, de los cuales el 20-40% serían APP. En nuestro país, existen varios estudios epidemiológicos en demencias8,9, estimándose la prevalencia de DFT en 0,2-0,3% en la población de mayores de 65 años10,11. Se ha sugerido una mayor prevalencia de APP en pacientes con trastornos del aprendizaje, como dislexia, lo que se explicaría por una mayor susceptibilidad de las redes neurales del lenguaje en estos pacientes o sus familias12. También se ha detectado una mayor frecuencia de vasectomía, lo que sugeriría una base autoinmunitaria13, aunque estas asociaciones no han sido replicadas.
La gran mayoría de casos de APP son esporádicos14,15. Una proporción de pacientes pertenecen a familias con mutaciones en el cromosoma 17, bien en el gen que codifica la proteína tau asociada a microtúbulos (MAPT) o la progranulina16,17. Los casos con patología tau, habitualmente en el contexto de una demencia frontotemporal con afectación conductual18, se asocian a mutaciones en MAPT, que suelen conllevar una ganancia de función tóxica; por su parte, las mutaciones en la progranulina se relacionan con patología de demencia frontotemporal con inmunorreactividad a ubiquitina (DFT-U). Las mutaciones en la progranulina típicamente se presentan como afasia no fluente o logopénica16,18,19, mientras que las mutaciones de MAPT principalmente como afasia no fluente o semántica, en este último caso habitualmente con alteración conductual asociada18,20. No obstante, las distintas mutaciones pueden manifestarse con fenotipos variados y lo habitual es el hallazgo de síndromes distintos (afasia progresiva, DFT-variante conductual) dentro de los miembros de una misma familia16. Se ha planteado, además, que las mutaciones de la progranulina podrían condicionar un trastorno del lenguaje distintivo, no encuadrable totalmente en los subtipos habituales19.
El genotipo de la ApoE no ha demostrado valor predictivo adicional a la descripción clínica en el diagnóstico de enfermedad de Alzheimer (EA) en la APP en el estudio con mayor tamaño muestral21,22. Aunque el alelo e4 se encuentra presente en un porcentaje mayor de casos en la variante logopénica que en el resto de las formas clínicas, la prevalencia es menor que en la EA típica22,23. Por su parte, el polimorfismo de MAPT H1/H1 se halla con más frecuencia en la APP esporádica24, sugiriéndose como factor de riesgo para degeneración corticobasal y parálisis supranuclear progresiva y, en menor medida, para demencia frontotemporal25–27. También los heterocigotos para el codón 129 de la proteína priónica podrían tener mayor riesgo28.
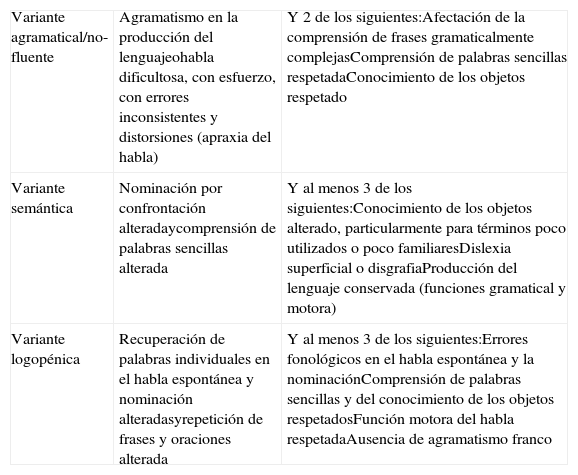
Tipos clínicosSe han descrito 3 subtipos clínicos principales de APP, que a su vez se asocian a un patrón radiológico anatómico y funcional3,29. Estas variantes son las formas agramatical/no fluente (APP-G); semántica (APP-S); y logopénica/fonológica (APP-L), incluyéndose las 2 primeras en el grupo de demencias frontotemporales. Existe, sin embargo, una minoría de pacientes («APP inclasificable») que no pueden incluirse en ninguno de estos grupos, bien porque presentan síntomas lingüísticos aislados o bien porque tienen características de más de un grupo30. Recientemente se ha publicado un consenso de criterios de diagnóstico y clasificación de la APP en sus 3 variantes principales30 (tabla 1). Este consenso, aunque pendiente de su validación definitiva y con el que no todos los autores están de acuerdo, pretende evitar la disparidad existente previamente entre los diferentes centros y estudios, que dificultaba la extrapolación de resultados.
Criterios de diagnóstico clínico de las tres variantes de APP
| Variante agramatical/no-fluente | Agramatismo en la producción del lenguajeohabla dificultosa, con esfuerzo, con errores inconsistentes y distorsiones (apraxia del habla) | Y 2 de los siguientes:Afectación de la comprensión de frases gramaticalmente complejasComprensión de palabras sencillas respetadaConocimiento de los objetos respetado |
| Variante semántica | Nominación por confrontación alteradaycomprensión de palabras sencillas alterada | Y al menos 3 de los siguientes:Conocimiento de los objetos alterado, particularmente para términos poco utilizados o poco familiaresDislexia superficial o disgrafiaProducción del lenguaje conservada (funciones gramatical y motora) |
| Variante logopénica | Recuperación de palabras individuales en el habla espontánea y nominación alteradasyrepetición de frases y oraciones alterada | Y al menos 3 de los siguientes:Errores fonológicos en el habla espontánea y la nominaciónComprensión de palabras sencillas y del conocimiento de los objetos respetadosFunción motora del habla respetadaAusencia de agramatismo franco |
Modificado de Gorno-Tempini et al.30
La variante agramatical se caracteriza por un lenguaje hipofluente y laborioso, con agramatismo, parafasias fonémicas, dificultad en la comprensión de estructuras gramaticales complejas y en ocasiones apraxia del habla. Puede haber acalculia, apraxia ideomotora y orofacial, todo ello de leve intensidad al menos inicialmente. También puede estar alterada la prosodia.
La variante semántica se produce por una pérdida progresiva del conocimiento semántico31, lo que se manifiesta clínicamente como un lenguaje fluente y gramaticalmente correcto, pero con alteración en la nominación por confrontación, en la comprensión de palabras sencillas y en el conocimiento de los objetos, con un discurso progresivamente vacío de significado, con circunloquios y parafasias semánticas. En cambio, la comprensión de frases y la repetición están conservadas. Puede haber, además, dislexia superficial y agnosia visual asociativa, así como prosopagnosia32. Mesulam considera que es conveniente distinguir entre los pacientes con un trastorno puramente lingüístico (APP-semántica) y aquellos que asocian agnosia visual u otros déficits (demencia semántica) de reconocimiento (agnosia táctil, gustativa, etc.)33,34. Esta opinión no es compartida por otros autores, quienes consideran que APP-S y demencia semántica son la misma entidad, probablemente en diferentes estadios evolutivos, pero con una anatomía patológica común35.
La variante logopénica se caracteriza por un lenguaje marcadamente anómico, con frecuentes pausas por este motivo. Esto produce un habla lenta y con una fluencia falsamente reducida, con frecuentes pausas durante la búsqueda de palabras, lo que podría confundirse con la variante agramatical23,36. Sin embargo, la articulación, la prosodia y la gramática están conservadas30,37, así como la comprensión de palabras sencillas. La repetición de frases, en cambio, está también afectada, siendo esta, junto con el déficit en la evocación de palabras, las 2 características fundamentales. Por todo ello, se asemeja con la afasia de conducción. A diferencia de la APP-semántica, en la afasia logopénica los pacientes suelen ser capaces de señalar el objeto correcto cuando este es nominado por el explorador y pueden describir (o al menos gesticular) su uso, ya que el trastorno parece ser secundario al daño en el circuito fonológico de la memoria de trabajo, conservando en cambio el significado de las palabras.
Aunque las variantes clínicas se definen por la semiología de la afasia, existen otros síntomas asociados que pueden ayudar en el diagnóstico. Así, las alteraciones conductuales similares a la demencia frontotemporal-variante conductual (DFT-vc) se encuentran con frecuencia en la variante semántica, pero no en la agramatical ni logopénica, especialmente desinhibición, comportamientos motores aberrantes y trastornos de la alimentación38. Los signos extrapiramidales en la exploración neurológica inicial son más frecuentes en las variantes agramatical y logopénica que en la semántica, en que son excepcionales31,39. En concreto, bradicinesia (hipomimia sobre todo) y rigidez (fundamentalmente apendicular y bilateral) son más frecuentes en la variante agramatical39, mientras que las alteraciones de la marcha (menos específicas de patología extrapiramidal) en la forma logopénica. Por tanto, las alteraciones conductuales y el parkinsonismo leve podrían tener valor en la distinción entre formas clínicas en casos dudosos, aunque no se contemplen entre los criterios diagnósticos.
Evolución y pronósticoDe forma análoga a la demencia tipo Alzheimer, se ha planteado el uso de una fase preclínica y prodrómica que podría llamarse «deterioro cognitivo leve afásico». Sin embargo, no está clara su utilidad. Los trastornos de memoria son frecuentes en el envejecimiento normal y, por tanto, es de utilidad distinguir entre normalidad y patología. En cambio, las alteraciones del lenguaje son siempre patológicas y no pueden por ello ser atribuidas únicamente al envejecimiento15.
La edad media de inicio suele situarse entre los 50-70 años40, aunque puede aparecer en un amplio rango de edad7. Una vez establecido, el trastorno afásico tiende a progresar, evolucionando en muchos casos hacia el mutismo40. La pérdida de la independencia ocurre de forma más tardía que en otras demencias (a los 7 años en el 50% de los pacientes según LeRhun et al.; la supervivencia, en cambio, es similar, en torno a los 7-10 años40, aunque estudios recientes sugieren un curso más lentamente progresivo41. En algunos, el lenguaje sigue siendo la única o predominante manifestación clínica. En otros, sin embargo, aparecen otros déficit adicionales (cognitivos, conductuales, extrapiramidales, etc.). Se desconoce, no obstante, con qué frecuencia los pacientes con APP desarrollan una demencia generalizada u otros déficit (denominada «APP plus»14,15) en el curso de la enfermedad. En fases iniciales, se admite la presencia de acalculia, apraxia ideomotora y visuoconstructiva, siempre que sean de intensidad leve y que no sean la causa de la limitación funcional del paciente. En el estudio de seguimiento longitudinal de Kertesz et al.42, un 54% desarrolló durante el seguimiento un segundo o tercer síndrome distinto de la APP (DFT, PSP, DCB).
La evolución puede ser monitorizada clínicamente, aunque puede ser complicado el uso de las escalas habituales utilizadas para la EA, dada la influencia del lenguaje sobre el resto de dominios cognitivos43. El test Addensbrook's Cognitive Examination puede ser útil en el diagnóstico y evolución44. Se ha diseñado además una escala específica, Progressive Aphasia Severity Scale (PASS)45), que evalúa 10 dominios del lenguaje tras la entrevista clínica con el paciente y cuidador, y se ha correlacionado con el grado de atrofia selectiva46.
Anatomía patológicaEl criterio temporal de 2 años dominados por la afasia intenta excluir a pacientes con enfermedad de Creutzfeldt-Jakob y formas típicas de EA con afectación del lenguaje de inicio. Sin embargo, aunque útil, este criterio, que por otra parte no ha sido claramente validado en estudios longitudinales47, no consigue simplificar la anatomía patológica en la APP, que es heterogénea. Las patologías subyacentes pueden clasificarse en 3 grandes grupos: taupatías del espectro de la degeneración frontotemporal, incluyendo enfermedad de Pick, PSP, DCB y otras menos frecuentes como taupatía de múltiples sistemas o demencia por cuerpos argirófilos; degeneración frontotemporal tau negativa y ubiquitina positiva (DFT-U), principalmente en relación con TDP-43; y EA. De forma menos frecuente se halla demencia sin histopatología distintiva o demencia con cuerpos de Lewy.
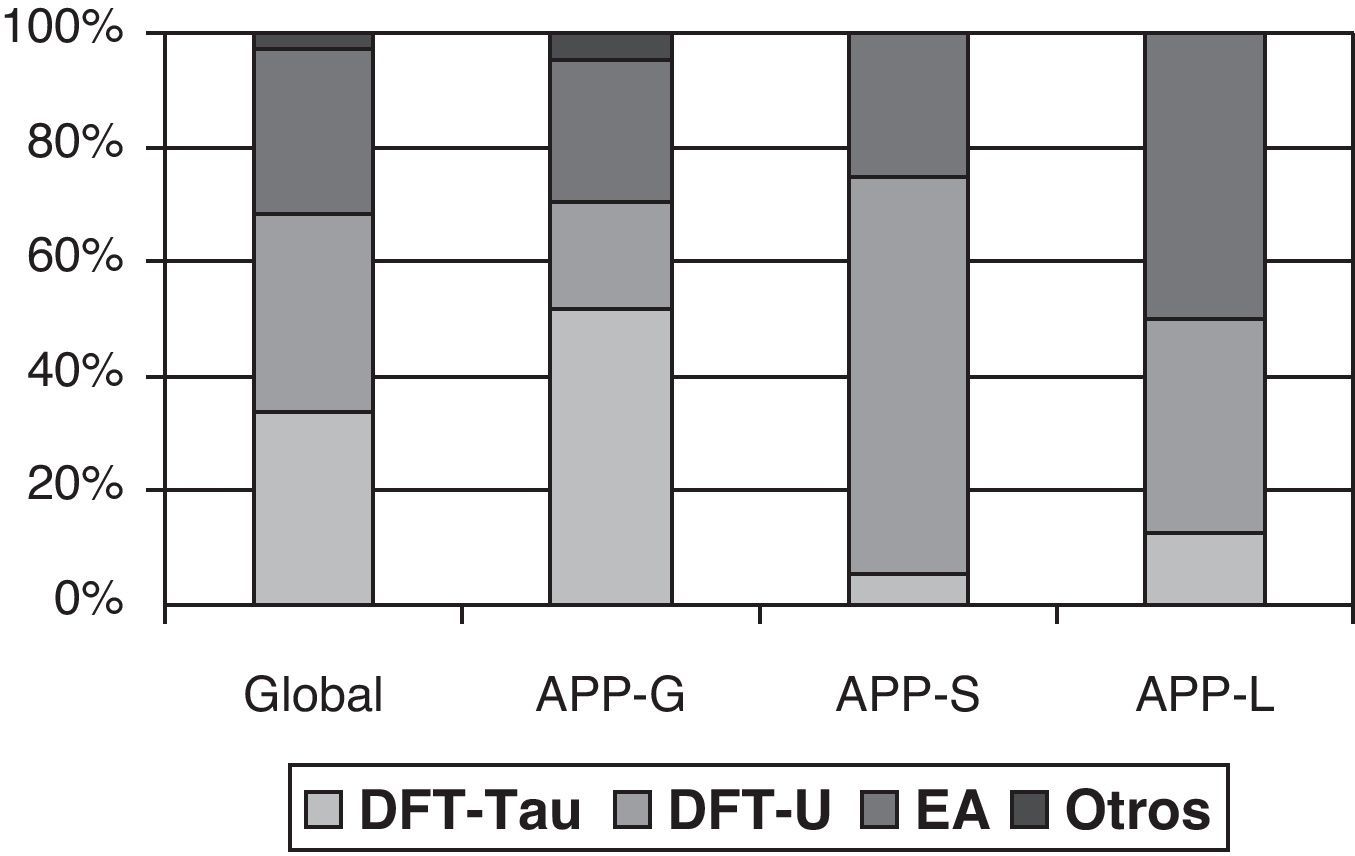
Los estudios realizados de correlación clínico-patológica han demostrado una asociación preferente de cada una de las formas clínicas con un grupo anatomopatológico. Estos estudios han sido revisados por Grossman7, incluyendo 145 casos de 7 series distintas, y demostrando una mayor asociación de afasia progresiva no fluente con DFT-tau positiva, demencia semántica con DFT-U y afasia logopénica con EA. Si bien la asociación de cada forma clínica con una patología concreta se estima entre el 50-70% de los casos, la distinción entre formas clínicas mejora la capacidad predictiva de la patología subyacente (fig. 1). La mayoría de los estudios, sin embargo, se centran en uno o dos subtipos de APP y los criterios diagnósticos de las diferentes formas clínicas no fueron iguales, mostrando en ocasiones resultados discordantes21,31,42,47–51. De hecho, algunos autores abogan por distinguir, dentro de la APP-agramatical, aquellos pacientes con apraxia del habla o disartria pura, quienes realmente se asociarían con patología tau, de aquellos con afasia agramatical, que se asociaría a DFT-ubiquitina50,52,53.

Anatomía patológica de la APP. Obtenido a partir de los datos de Grossman7.
Se ha tratado de correlacionar ciertos hallazgos clínicos, neuropsicológicos y complementarios con la patología subyacente, como se resumen en la tabla 2. Así, la apraxia del habla o los signos extrapiramidales se asociarían a DFT-tau, mientras que la alteración de memoria episódica precoz parece estar relacionada con EA. La aparición de signos de motoneurona es probablemente el hallazgo más específico, indicando DFT-U. También se han evaluado los datos demográficos, como edad de presentación y supervivencia. Se ha encontrado una tendencia a un inicio más precoz de los casos con DFT-tau negativa42,54 respecto de DFT-tau y EA, si bien este dato no ha sido contrastado en otros estudios21,31,55 y podría estar influido por el subgrupo de pacientes con enfermedad de motoneurona. La supervivencia tampoco es distinta entre los diferentes grupos. Aunque la predicción de la anatomía patológica a partir de datos individuales puede conducir a error21, la consideración conjunta de la clínica, neuropsicología y neuroimagen (anatómica o funcional) puede incrementar las posibilidades de acierto56. No obstante, faltan estudios que validen este abordaje en la APP.
Manifestaciones y hallazgos que podrían predecir una determinada anatomía patológica en la APP
| Manifestaciones o hallazgos | Anatomía patológica que sugiere |
| Clínicos | |
| Apraxia del habla asociada a afasia | Taupatía (en concreto, DCB)49 |
| Apraxia del habla predominante o aislada | Taupatía (especialmente PSP)49 |
| Parálisis supranuclear de la mirada vertical, enlentecimiento de sacadas verticales | PSP49 |
| Alteración conductual precoz en la variante semántica | DFT (hace poco probable EA)31,54 |
| Signos de enfermedad de motoneurona | DFT-ubiquitina31 |
| Signos extrapiramidales | Taupatía39,55,100 |
| Neuropsicológicos | |
| Pérdida de memoria precoz, afectación de memoria visual | EA42,62 |
| APP-logopénica con baja puntuación en tests de memoria | Enfermedad de Alzheimer21,42 |
| Afectación ejecutiva (construcción visual, digit span inverso) | Taupatía (comparado con EA y TDP-43)62,100,101 |
| Mayor afectación de fluencia verbal fonémica (Controlled Oral Word Association Test) | DFT-U respecto a EA62 |
| De imagen | |
| Atrofia e hipoperfusión o hipometabolismo temporoparietal (especialmente bilateral) en variante hipofluente. | EA68 |
| Atrofia temporoparietal y frontal lateral izquierdos, con respeto de temporal anterior | EA (en lugar de DFT-U)62 |
| Atrofia frontal medial y temporal anterior izquierdos, con respeto de parietal | DFT-U (en lugar de EA)62 |
Un tema controvertido es el papel de la EA en la APP57: aunque presente en muchas de las autopsias, algunos autores dudan de su papel causal21,58,59. Si bien el patrón de atrofia predomina en las áreas del lenguaje, la distribución de ovillos neurofibrilares y placas neuríticas no es diferente de la de los pacientes con EA que presentan un síndrome amnésico típico, y tampoco existe asimetría entre los dos hemisferios cerebrales. Es decir, no existen claramente más lesiones características de EA en las áreas que justificarían su presentación clínica con un trastorno afásico, aunque en este punto no todos los estudios coinciden47,60. En cualquier caso, esto plantea la posibilidad de que la EA no sea la causa real de la sintomatología de los pacientes, pudiendo coexistir junto a otra enfermedad neurodegenerativa en un mismo paciente, tal vez por concurrencia de causas o factores etiopatogénicos. La EA podría, de acuerdo con esta hipótesis, enmascarar la verdadera anatomía patológica subyacente en la APP. A este respecto, la demostración de argyrophilic thorny astrocyte clusters en pacientes con APP con EA, pero no en pacientes con presentación típica de EA58, apoyarían la hipótesis de una concurrencia de procesos neurodegenerativos. Este hallazgo, sin embargo, no ha podido ser contrastado en un estudio posterior59. También se ha evaluado con un objetivo similar la presencia de inclusiones TDP-43, sospechándose la presencia de anatomía patológica de DFT encubierta en los casos de EA, encontrándose, sin embargo, en un bajo porcentaje de los casos de APP con EA59. Si, por el contrario, se trata de un inicio focal de la EA, como coinciden en señalar gran parte de los autores51,57, se desconoce por qué adopta este patrón de inicio o si la respuesta al tratamiento anticolinesterásico es equivalente a la EA típica, entre otras preguntas.
Neuroimagen anatómica y funcionalLa resonancia magnética ha sido utilizada en distintos estudios a fin de describir los patrones de atrofia asociados a cada una de las variantes con la evaluación del grosor cortical. En líneas generales, se halla atrofia frontal inferior e insular izquierda en la APP-agramatical, temporal anterior bilateral de predominio izquierdo en la variante semántica, y temporoparietal izquierda en la forma logopénica3 (tabla 3). Con la evolución, la atrofia se extendería a otras zonas, también con un patrón diferente según cada forma clínica61. Ciertos patrones de atrofia, además, podrían ser predictivos de la anatomía patológica subyacente62 o de la evolución clínica63. Últimamente, se han realizado, asimismo, estudios de tractografía con tensor de difusión64–67, observándose la afectación de distintas vías del lenguaje en cada variante. En la forma no-fluente se observa una afectación de las vías dorsales (fascículo longitudinal superior), mientras que en la afasia logopénica se daña principalmente el componente temporoparietal de la vía dorsal. Estos estudios ponen de manifiesto que en la APP se produce también un daño de la sustancia blanca y de las diferentes redes neuronales que participan en el lenguaje64,66; pueden ser útiles, asimismo, en la descripción del funcionamiento de las vías involucradas en el lenguaje y es posible que ciertas vías puedan asociarse a determinadas patologías si, como se ha demostrado, ciertos síntomas lingüísticos podrían ser predictores de ellas49.
Patrón de atrofia en RM
| APP-agramatical | Frontal inferior e ínsula izquierdas.Si existe mutismo: pars opercularis y ganglios basales izquierdos. |
| APP-semántica | Temporal anterior bilateral de predominio izquierdo |
| APP-logopénica | Temporal posterior y parietal inferior izquierdo. |
| Apraxia del habla | Áreas premotora y motora suplementaria izquierdas |
Los estudios realizados con tomografía por emisión de positrones (PET), algunos de ellos con correlación patológica68, han demostrado que la captación es focal en todas las afasias progresivas, independientemente de la anatomía patológica subyacente69–71. El hipometabolismo es prerrolándico en las formas no-fluentes y posrolándico en las fluentes72. Existen además distintos patrones asociados a las 3 formas clínicas principales: hipometabolismo frontal (APP-agramatical), temporal anterior (APP-semántica) y temporoparietal (APP-logopénica), siempre de predominio izquierdo71. El hipometabolismo temporoparietal, especialmente bilateral, sugiere anatomía patológica de EA68. También se ha utilizado PET con ligando Pittsburgh, encontrándose aumentado más frecuentemente en la forma logopénica, lo que sería congruente con su mayor asociación a EA71. Los estudios realizados parecen demostrar una mayor sensibilidad de la PET respecto a SPECT y RM anatómica73, un patrón focal asociado a cada una de las variantes71,72 y una capacidad de determinados patrones de predecir la anatomía patológica68. Sin embargo, el tamaño muestral de los estudios es pequeño y, por tanto, las conclusiones están pendientes de validación.
Biomarcadores en el líquido cefalorraquídeoLos biomarcadores en el LCR han sido ampliamente estudiados en la demencia tipo Alzheimer, bien para confirmar el diagnóstico, o bien para identificar a pacientes con mayor riesgo en fases prodrómicas74,75. También se enfatiza su uso como evaluadores de la eficacia de fármacos modificadores de la enfermedad76. En los estudios realizados, el patrón de aumento de tau (total y fosforilada) y disminución de A-Beta-1-42 es una herramienta diagnóstica sensible y específica en el diagnóstico de EA. Estos biomarcadores también podrían utilizarse para el diagnóstico de formas de inicio atípicas de enfermedad de Alzheimer, incluyendo la afasia progresiva, ayudando así a discriminar de aquellas formas con otra patología subyacente77. La DFT se asociaría a un patrón inverso a la enfermedad de Alzheimer, con tau disminuida y A-beta-1-42 normal o aumentada78. La ratio tau/A-beta-42 puede ser especialmente sensible y específica en discriminar EA y DFT79,80. Por tanto, los biomarcadores en el LCR pueden utilizarse en la APP para el diagnóstico de la patología subyacente. Sin embargo, no se han realizado estudios en series amplias de pacientes con APP y, por ello, no hay evidencia de la utilidad de estos biomarcadores en la identificación de pacientes con anatomía patológica de enfermedad de Alzheimer en este subgrupo de pacientes81–83. Faltan, además, marcadores específicos de degeneración frontotemporal y de sus diferentes formas (taupatías, ubiquitinapatías) que permitan un diagnóstico patológico in vivo o, al menos, aumenten la capacidad predictiva de la clínica y los otros estudios mencionados previamente. Algunos de los propuestos han sido, entre otros, cromogranina B, cistatina C e IL-1784. Todo ello será de gran utilidad con la aparición de los nuevos tratamientos modificadores de la vía de betaamiloide y tau.
TratamientosEl tratamiento se centra en 3 pilares básicos: tratamientos farmacológicos sintomáticos y/o modificadores del curso de la enfermedad; rehabilitación del lenguaje y estrategias para mejorar la comunicación; y ayudas a paciente y cuidadores85.
No existen, por el momento, ensayos clínicos a gran escala y los estudios disponibles, de pequeño tamaño muestral y en ocasiones en el seno de ensayos en pacientes con demencia frontotemporal, no han demostrado eficacia de ningún tratamiento farmacológico. Se han probado en ensayos piloto bromocriptina86, galantamina87, rivastigmina88, selegilina89 y memantina90,91. Tan solo galantamina pareció demostrar cierta tendencia no significativa a la estabilización de la afasia. También se han sugerido otros tratamientos, como corticoides92, pero con experiencias limitadas a casos individuales93. Aunque no existen estudios con anticolinesterásicos en la variante logopénica, parece oportuno intentar su uso en casos en que exista sospecha de EA subyacente. Por otra parte, debido a que muchos pacientes conservan la consciencia de enfermedad durante años, son convenientes la evaluación y el tratamiento de la depresión en caso de estar presente94.
Tampoco existen estudios que hayan evaluado la utilidad de la logoterapia, aunque esta se supone útil, habiéndose demostrado mejoría clínica en casos individuales, a través de la activación de áreas corticales preservadas durante la rehabilitación en las 3 variantes clínicas95–99. Algunos pacientes también pueden aprender formas de comunicación alternativa33.
ConclusionesLa APP es un síndrome clínico con 3 variantes semiológicas principales, con una correspondencia anatómica concreta. Es una entidad cada vez más diagnosticada, conforme se avanza en su conocimiento y se constata su utilidad clínica en el ámbito de las enfermedades neurodegenerativas. Existe, además, un número significativo de casos con APP que puede clasificarse erróneamente dentro de la demencia tipo Alzheimer, dada la evaluación de la memoria verbal (pero no de otras modalidades, como la memoria visual) en algunos de los tests neuropsicológicos más utilizados, y el amplio uso del síntoma «no encontrar palabras» para reflejar un problema mnésico y/o lingüístico. Puede haber, además, pacientes con enfermedad de Alzheimer típica que asocian desde el inicio una marcada alteración del lenguaje, o pacientes con APP que desde fases precoces presentan una importante pérdida de memoria. Éstas pueden ser formas de solapamiento, que probablemente estén más próximas a la EA que al espectro de la degeneración frontotemporal.
La APP es un buen ejemplo, asimismo, de las posibilidades y limitaciones del diagnóstico sindrómico, ya que una mejor descripción clínica ha permitido identificar 3 variantes bien definidas, cada una de ellas con una asociación anatomopatológica preferencial, si bien no completa. Por ello, es necesario el perfeccionamiento de los distintos biomarcadores (imagen anatómica y funcional, proteínas en el LCR) y, probablemente, también seguir profundizando en la descripción clínica y neuropsicológica, con el fin de lograr una aproximación al diagnóstico anatomopatológico durante la vida del paciente. De hecho, en los últimos años, la mejoría en el diagnóstico ha sido mayor por el perfeccionamiento de la descripción semiológica que por el desarrollo de técnicas complementarias analíticas o de imagen. De cualquier modo, todo ello debería permitir el uso adecuado de tratamientos modificadores de la enfermedad cuando estos estén disponibles, lo que tendría un impacto favorable sobre estos pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

