



En el cerebro del humano y en el de modelos animales, la enfermedad de Alzheimer (EA) se caracteriza por la acumulación del péptido β-amiloide (βA), de la proteína tau hiperfosforilada, degeneración neuronal y gliosis astrocítica que son prominentes en regiones cerebrales vulnerables (hipocampo y corteza). Estas alteraciones se relacionan con el deterioro cognitivo (pérdida de la memoria) y no cognitivo en la función motora. El objetivo de este trabajo fue identificar en el modelo (3xTg-AD) los cambios celulares (neuronas y astroglía) y la agregación de βA y tau hiperfosforilada en la corteza motora primaria (M1) en una etapa intermedia de la EA y su relación con el desempeño motor.
MétodosSe utilizaron hembras 3xTg-AD de 11 meses de edad, comparadas con no transgénicas (No-Tg) de la misma edad. En ambos grupos, se evaluaron el desempeño motor (campo abierto) y el daño celular con marcadores específicos: BAM10 (agregados βA extracelulares), tau 499 (hiperfosforilada), GFAP (astrocitos) y Klüver-Barrera (neuronas) en la M1.
ResultadosLas hembras 3xTg-AD en etapa intermedia de la patología mostraron alteraciones motoras y celulares asociadas al depósito de βA y tau hiperfosforilada en la M1.
ConclusionesDesde etapas tempranas de la EA se observan signos y síntomas de deterioro funcional. Sin embargo, en este estudio reportamos que en la etapa intermedia de la patología se encuentran establecidas las características de daño en la M1 asociadas al desempeño motor. Eventos que se relacionan con el avance de las características clínicas de la patología.
In humans and animal models, Alzheimer disease (AD) is characterised by accumulation of amyloid-β peptide (Aβ) and hyperphosphorylated tau protein, neuronal degeneration, and astrocytic gliosis, especially in vulnerable brain regions (hippocampus and cortex). These alterations are associated with cognitive impairment (loss of memory) and non-cognitive impairment (motor impairment). The purpose of this study was to identify cell changes (neurons and glial cells) and aggregation of Aβ and hyperphosphorylated tau protein in the primary motor cortex (M1) in 3xTg-AD mouse models at an intermediate stage of AD.
MethodsWe used female 3xTg-AD mice aged 11 months and compared them to non-transgenic mice of the same age. In both groups, we assessed motor performance (open field test) and neuronal damage in M1 using specific markers: BAM10 (extracellular Aβ aggregates), tau 499 (hyperphosphorylated tau protein), GFAP (astrocytes), and Klüver-Barrera staining (neurons).
ResultsFemale 3xTg-AD mice in intermediate stages of the disease displayed motor and cellular alterations associated with Aβ and hyperphosphorylated tau protein deposition in M1.
ConclusionsPatients with AD display signs and symptoms of functional impairment from early stages. According to our results, M1 cell damage in intermediate-stage AD affects motor function, which is linked to progression of the disease.
La demencia es un síndrome que se caracteriza por una disminución significativa del rendimiento cognitivo (amnesia) y alteraciones psicológicas y de las funciones ejecutivas con repercusión en las actividades cotidianas y en la autonomía del paciente1. La enfermedad de Alzheimer (EA) es la causa más común de demencia en adultos mayores; es una enfermedad heterogénea, se acompaña de manifestaciones clínicas, como desorden cognitivo, pérdida de la memoria y cambio del comportamiento2, y no cognitivas, como la disfunción motora con disturbios en el balance, disminución en la velocidad del caminado y signos motores generales que puede representase como pérdida de la masa y fuerza muscular que de manera progresiva provocan la pérdida de la independencia3-5; estos eventos se proponen como una característica preclínica de la EA6,7. Aunque el predictor de demencia es la disminución del volumen del hipocampo, también existen formas de inicio focal o asimétrico como la atrofia cortical que representa los síntomas no cognitivos de la patología como apraxias progresivas y compromiso cerebral asimétrico8. En la evaluación clínica se distinguen 3 etapas: la hipocámpica (con el defecto amnésico), seguida de trastornos del lenguaje y apraxias (alteración funcional de las áreas corticales de asociación, parietal, temporal y occipital), y la global, que incluye la vía extrapiramidal8.
El mecanismo celular concomitante en la EA es la disfunción sináptica que se inicia en las áreas vulnerables (hipocampo y la neocorteza) en donde ocurre la acumulación del péptido β-amiloide (βA) y la hiperfosforilación de la proteína tau del citoesqueleto9,10. Estos eventos producen la neurodegeneración de tipo irreversible, aunado a la pérdida progresiva de la memoria11. El βA es un péptido cuyas funciones fisiológicas está la de controlar el transporte del colesterol12, pero a medida que se acumula en oligómeros de 32 a 42 aminoácidos puede ocasionar alteraciones sinápticas y daño neuronal13. Otros factores asociados a la patología son el estrés oxidante, la neuroinflamación que provoca la presencia de las placas βA y la truncación e hiperfosforilación de la proteína tau que produce cambios del citoesqueleto de las neuronas del hipocampo14, sitio de plasticidad neuronal, que regula la memoria y el aprendizaje15. En ratones transgénicos este déficit de la memoria y la disfunción neuronal inducida por βA se recupera cuando se reduce la citotoxicidad de tau16.
Actualmente, existen diversos modelos de ratones para el estudio progresivo de la EA17 pero es el triple transgénico (3xTg-AD) el que sobre expresa las 3 proteínas humanas (PS1M146V APPSwe y tauP3001) y recapitula gran parte de los aspectos del desarrollo progresivo de la EA, porque muestran depósitos de βA (placas) y tau hiperfosforilada, dependiendo de la edad y de las regiones cerebrales específicas. Al mismo tiempo presentan déficit cognitivo relacionado con placas de βA, entre los 3 y 4 meses de edad en la región CA1 del hipocampo y los depósitos extracelulares son evidentes de los 6 a los 12 meses en la corteza frontal18. Características que permiten investigar varias alternativas de posible tratamiento, en etapas intermedias de la manifestación de la EA. Además, durante su progresión, se activa la microglía y se produce la astrogliosis reactiva en el interior de las placas amiloides19; de manera particular se activan los astrocitos, en respuesta al daño, incrementando la expresión de la proteína fibrilar acídica glial (GFAP) en sus filamentos intermedios, además se observan cambios morfológicos como hipertrofia20,21. Los astrocitos de manera dinámica proveen a las neuronas de soporte trófico y metabólico, modulan el procesamiento de la información, regulan la actividad neuronal y la plasticidad sináptica22-24. La desregulación de la homeostasis mantenida por los astrocitos25 puede tener graves consecuencias para la estabilidad de los microcircuitos neuronales y en la EA, es concomitante con el depósito extracelular de las proteínas βA y tau hiperfosforilada26. El objetivo del presente trabajo fue evaluar en la corteza motora primaria (M1) de control motor, los cambios ocurridos (en una etapa intermedia, 11 meses) antes del depósito de las placas y su posible relación con el aspecto no cognitivo (deficiencias motoras) de esta patología en el ratón transgénico 3xTg-AD que sobreexpresa las 3 proteínas humanas observadas en la EA.
MétodosAnimalesTodos los animales utilizados para el presente estudio fueron mantenidos y supervisados por un veterinario calificado, conforme a las normas internacionales para el manejo y uso de animales de experimentación establecidas por los National Institutes of Health (NIH) y la National Academy of Science y aprobado por el comité de Bioética del Instituto de Neurobiología de la Universidad Nacional Autónoma de México. Fueron mantenidos 4 animales por caja con acceso libre al agua y comida así como en óptimas condiciones de vivarium (12-h: 12 h del ciclo luz/obscuridad, temperatura 20°C, y humedad relativa en el ambiente del 40-50%). El modelo del ratón triple transgénico (3xTg-AD) posee 3 transgenes humanos (APPSwe y tauP301L sobre un mutante PS1M146V knock-in) y fue desarollado en el laboratorio del Dr. La Ferla (Universidad de California, Irvine)17, en el fondo híbrido de ratones control (129/C57BL6) y los no transgénicos (No-Tg) del mismo fondo híbrido que el control. Solo se utilizaron ratones hembra de 11 meses de edad; estos híbridos de las unidades reproductivas originales fueron destetados a los 30 días de edad. Los ratones del grupo 3xTg-AD eran homocigotos para la EA y no los controles (No-Tg); ambos fueron alojados 3 animales por jaula habitación, desde el destete hasta el inicio del experimento.
GenotipificaciónEn todos los animales utilizados, se realizó la extracción de ADN del segmento más caudal de la cola (0,5cm de largo) colocados en un tubo Eppendorf® de 1ml para realizar una lisis alcalina y, posteriormente, realizar la reacción en cadena de la polimerasa para comprobar la presencia de los genes de la proteína precursora amiloide y de la proteína tau (observadas las bandas a 500-bp y 350-bp, respectivamente), así como del gen de la presenilina 1 (PS1), en donde se observaron 2 bandas de 180 pb y 350 pb. Todos los geles fueron visualizados por luz UV.
Estudio conductualEl campo abierto es una prueba motora que es utilizada para determinar los niveles de actividad locomotora espontánea y los hábitos de exploración en el modelo de roedores con trastornos del sistema nervioso central17. En este estudio, se llevó a cabo en 8 animales por grupo y cada uno se colocó en una arena rectangular (45cm×45cm) durante 15 min con el experimentador fuera de su punto de vista. El suelo uniformemente iluminado fue marcado para visualizar la separación entre las áreas periféricas y las centrales, el movimiento se capturó con una cámara digital montada directamente encima del rectángulo. Las imágenes fueron trasmitidas al ordenador y grabadas con el seguimiento de la trayectoria de cada animal con el Advanced video-tracking Any maze (version 4.82) (Stoelting Co, EE. UU.) software, considerando como medida de la actividad locomotora la distancia en centímetros y el tiempo.
Fijación y procesamiento histológicoSeis animales por cada grupo fueron aleatoriamente seleccionados de los 16 empleados para la estudio conductual. Los animales fueron anestesiados i.p. con pentobarbital (300μg/kg de peso corporal), la perfusión intracardíaca se realizó con 4% de paraformaldehído amortiguado (pH 7,4, 0,1M) y 1.000 U del 0,1% de heparina y procaína. Los cerebros se retiraron del cráneo y se fijaron en 4% de paraformaldehído por 24 h a temperatura ambiente; se separaron los 2 hemisferios y cada uno se cortó sagitalmente a 50μm de espesor en un vibratomo (Vibratome 3000 tissue sectioning system Pelco, series 3000 Deluxe), se consideró entre las coordenadas de Bregma 1,92-2,28mm de acuerdo con el atlas del cerebro del ratón abarcando el punto lateral27. Un hemisferio se procesó con la técnica convencional de Klüver-Barrera para el análisis morfométrico de neuronas, que se visualizaron en un microscopio fotónico marca Nikon H550S, eclipse Ci, equipado con un DS-U2 S cámara y objetivo 100× planapocromático (Plan-Apochromat®, 1,25 NA/160), en cortes equidistantes y equivalentes para ambos grupos experimentales (3xTg-AD y No-Tg). Las mediciones se efectuaron en un área de 136×581μm de la corteza M1, con programas de software Q-Win e ImageJ (NIH, EE. UU.), para cuantificar las neuronas normales y las que mostraban daño. El criterio de inclusión para ser consideradas neuronas dañadas incluyeron: hipercromatismo citoplasmático, encogimiento celular y fragmentación nuclear; estas se expresaron como el número de neuronas por área determinada que de manera constante y para propósitos de comparación a nivel intersticial se expresaron como número de neuronas en un área de 0,1mm2. Para esto fue empleado el software Q-win e Image J (1.49; 1.7.0-95 NIH, EE. UU.).
Inmunohistoquímica para Aβ y tau hiperfosforiladaLa segunda serie de tejidos de cortes sagitales seriados de ambos hemisferios cerebrales de 50μm de espesor de la región M1, se utilizó para llevar a cabo las técnicas de inmunohistoquímica con tejidos flotantes (n=6 por cada grupo). Para anti-βA de ratón se usó Bam10 (IgG monoclonal, Sigma-Aldrich, No. A5213, 1: 1000) como anticuerpo primario y un anticuerpo secundario biotinilado (IgG anti-ratón, Vector: 1:500). Para teñir la proteína tau humana, se utilizó el anticuerpo primario Anti-Human PHF-Tau (IgG monoclonal, Thermo Scientific, 1:200), y un anticuerpo secundario biotinilado (IgG anti-ratón, Vector, 1: 500). Los kits de substrato de ABC y peroxidasa (Vector) se usaron para visualizar los anticuerpos unidos. Las imágenes se digitalizaron con una cámara DS-U2 S (Nikon Digital sight DS-V3) con un objetivo de 40X (Plan-Apochromat®, 0,65 NA/160).
Análisis inmunohistoquímico de astrocitosPara la identificación por fluorescencia de la GFAP, los tejidos se incubaron en PBS 0,1M con tritón al 0,1% por media hora y posteriormente se incubaron en PBS 0,1M con suero normal de cabra al 10% en Tritón 0,1% durante una hora en agitación a temperatura ambiente; después se retiró la solución y se incubó con el anticuerpo primario anti-GFAP (anticuerpo policlonal, hecho en conejo marca Dako con una concentración de dilución 1:500) toda la noche en agitación a 4°C. Después de 24h en incubación con el anticuerpo primario, este se retiró y se realizaron 5 lavados de 8min c/uno; posteriormente se incubó con el anticuerpo secundario asociado al fluorocromo lexa 488 marca Invitrogen con una dilución 1:500), se mantuvieron durante 2h a temperatura ambiente en oscuridad y con agitación continua, se hicieron 5 lavados de 8min c/u con PBS 0,1M y se montaron en porta objetos con Vectashield para mantener la fluorescencia y se sellaron con esmalte.
Para la identificación y la toma de imágenes de los tejidos en los que se evaluó la inmunorreactividad, se utilizó un microscopio confocal (marca Leica TCS SP2) en donde se obtuvieron las imágenes con el objetivo 100X planapocromático (Plan-Apochromat® 2,25 NA/160) y la cuantificación de células positivas para GFAP, en la M1 correspondió a un área de 136×581μm, la que se expresó como unidades arbitrarias de intensidad en un área de 0,1mm2 de manera constante, para lo cual se utilizó el programa ImageJ (National Institutes of Health, EE. UU.).
En el análisis de morfometría se consideró medir los cambios morfológicos de las prolongaciones de los astrocitos con una versión modificada del circulo de Sholl (1953)28, el cual consistió de 6 círculos concéntricos con una distancia de 10μm entre ellos, se sobrepuso el círculo central en el centro del astrocito, de forma que las prolongaciones celulares se observan intersectadas por los círculos concéntricos, las cuales fueron cuantificadas. Se eligieron de la M1, todos los astrocitos completos de cada hemisferio cerebral, en ambos grupos experimentales (3xTg-AD y No-Tg); este análisis permitió observar de manera comparativa, la morfología celular a través de la inmunorreactividad de los astrocitos.
Análisis estadísticoLa prueba conductual del campo abierto se analizó estadísticamente mediante la prueba de la t de Student y los resultados obtenidos de las cuantificaciones morfométricas e inmunohistoquímicas se analizaron mediante las pruebas t de Student y ANOVA para cada condición experimental con el empleo de los programas statView y Gradphad, y se expresaron con el error estándar de la media (SEM) con el promedio de 6 experimentos por grupo; los valores estadísticamente significativos se consideraron aquellos con un valor de p ≤ 0,05.
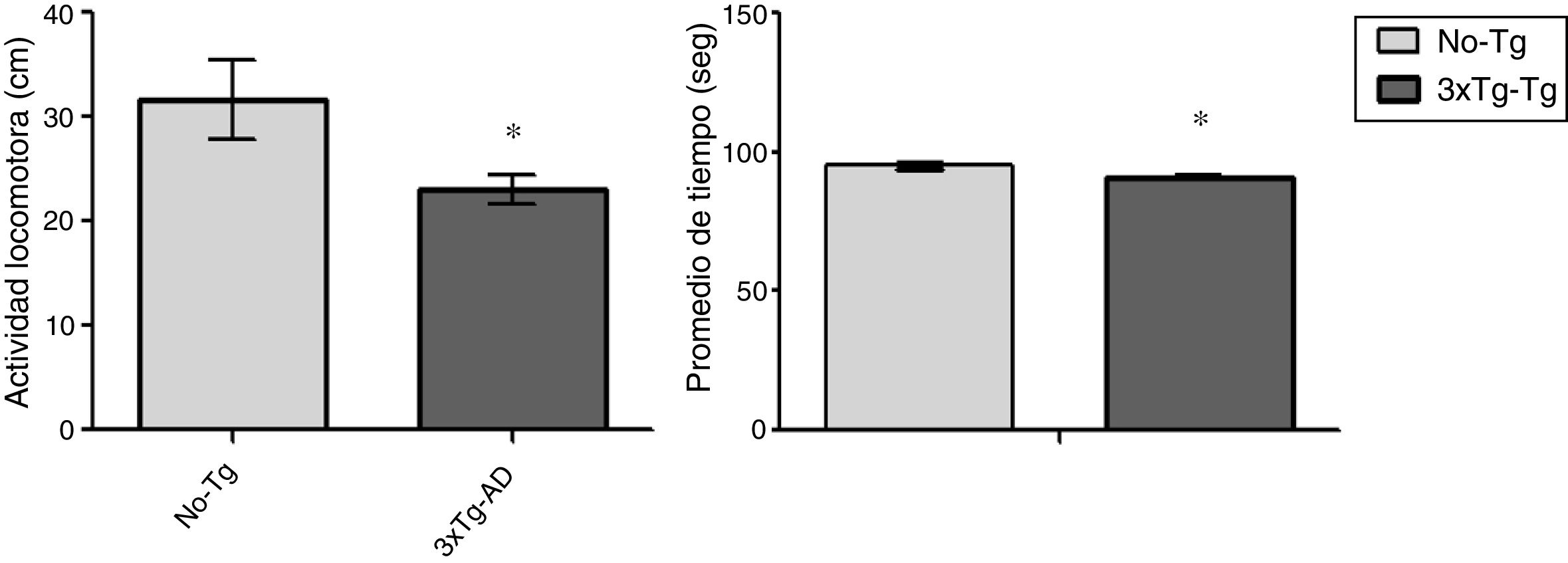
ResultadosCampo abiertoLa actividad locomotora fue significativamente (p<0,05) menor en los ratones 3xTg-AD que en los No-Tg durante la prueba de 15 min. La actividad motora se midió en los centímetros recorridos en el campo abierto y los ratones 3xTg-AD recorrieron menos distancia que el grupo No-Tg con menor tiempo en movimiento. El tiempo de inmovilidad no fue considerado en este análisis (fig. 1).
Análisis anatómico de corteza motora primaria y el tiempo (s) en la prueba de campo abierto por ambos grupos de ratones (No-Tg y 3xTg-AD). Se observó reducción significativa (p<0,05) en ambos parámetros del 3xTg-AD en comparación con los No-Tg.")
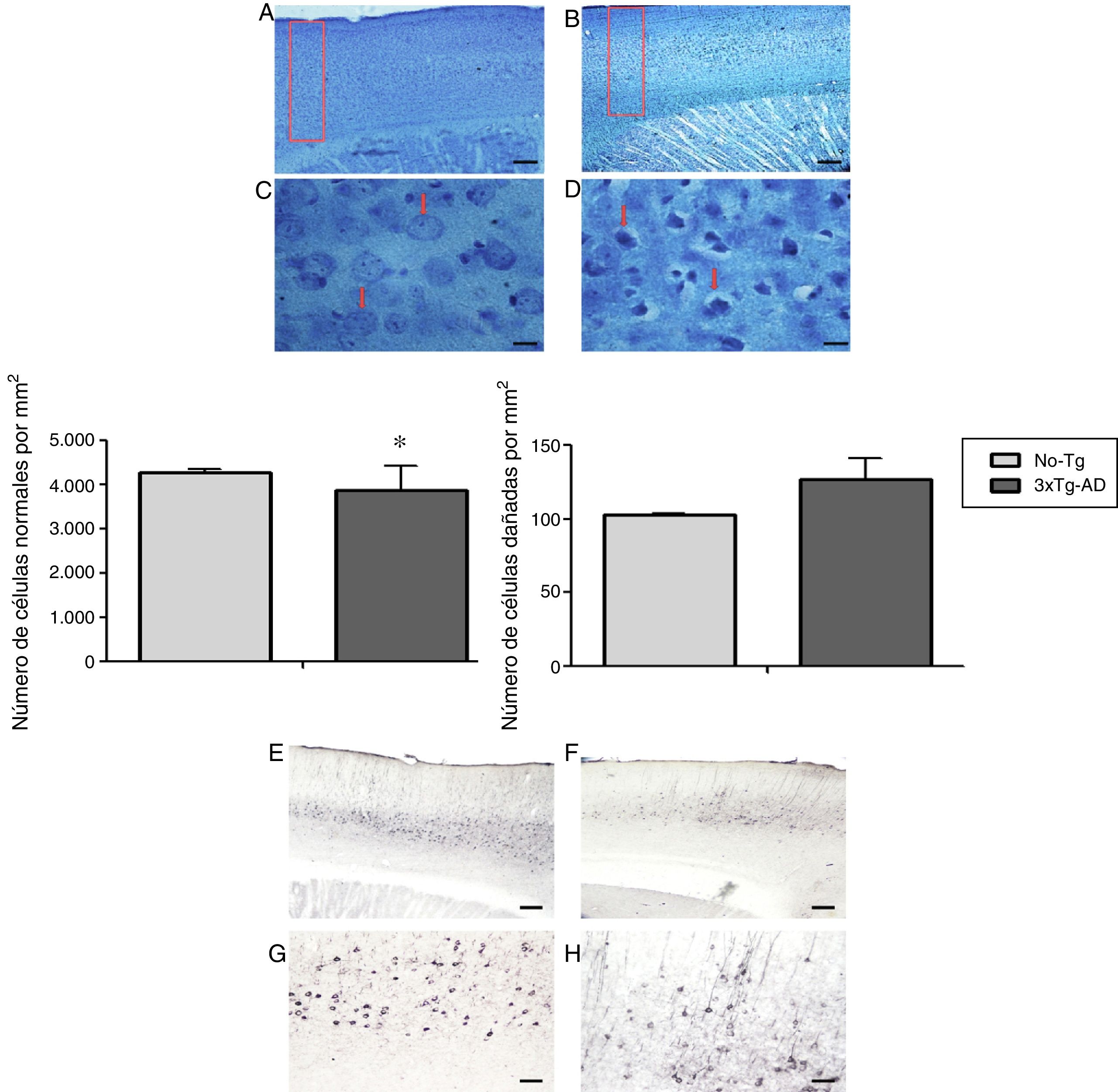
En el análisis morfométrico de las neuronas de la M1 del ratón 3xTg-AD (de 11 meses de edad, etapa intermedia de la enfermedad) se observó disminución significativa del número de neuronas normales, y un aumento en las anormales, que no presenta diferencias significativas (fig. 2) en los cortes teñidos con la técnica de Klüver-Barrera. En la misma área de la M1, las observaciones cualitativas analizadas con los marcadores específicos (BAM10) indica agregados beta amiloides extracelulares y tau 499, en su forma hiperfosforilada (fig. 2).
 del modelo 3xTg-AD de 11 meses de edad. Los cortes sagitales teñidos con la técnica de Klüver-Barrera en A y C; las flechas señalan a las células con características morfológicas normales (No-Tg). B y D) Tejido representativo del 3xTg-AD; las flechas señalan a las células con características de deterioro. Gráficas del promedio±EEM de células normales (con características de daño (n=6) con una significación p<0,05 (*). E y G) Corteza motora M1 del ratón triple transgénico (3xTg-AD) con el péptido β-amiloide (anticuerpo BAM-10). F y H) La tau con el anticuerpo 499. Barras de calibración: a 100 micras (A, B, E y F) y 10 micras (C, D, G y H).")
Fotomicrografías representativas de la corteza cerebral primaria (M1) del modelo 3xTg-AD de 11 meses de edad. Los cortes sagitales teñidos con la técnica de Klüver-Barrera en A y C; las flechas señalan a las células con características morfológicas normales (No-Tg). B y D) Tejido representativo del 3xTg-AD; las flechas señalan a las células con características de deterioro. Gráficas del promedio±EEM de células normales (con características de daño (n=6) con una significación p<0,05 (*). E y G) Corteza motora M1 del ratón triple transgénico (3xTg-AD) con el péptido β-amiloide (anticuerpo BAM-10). F y H) La tau con el anticuerpo 499. Barras de calibración: a 100 micras (A, B, E y F) y 10 micras (C, D, G y H).
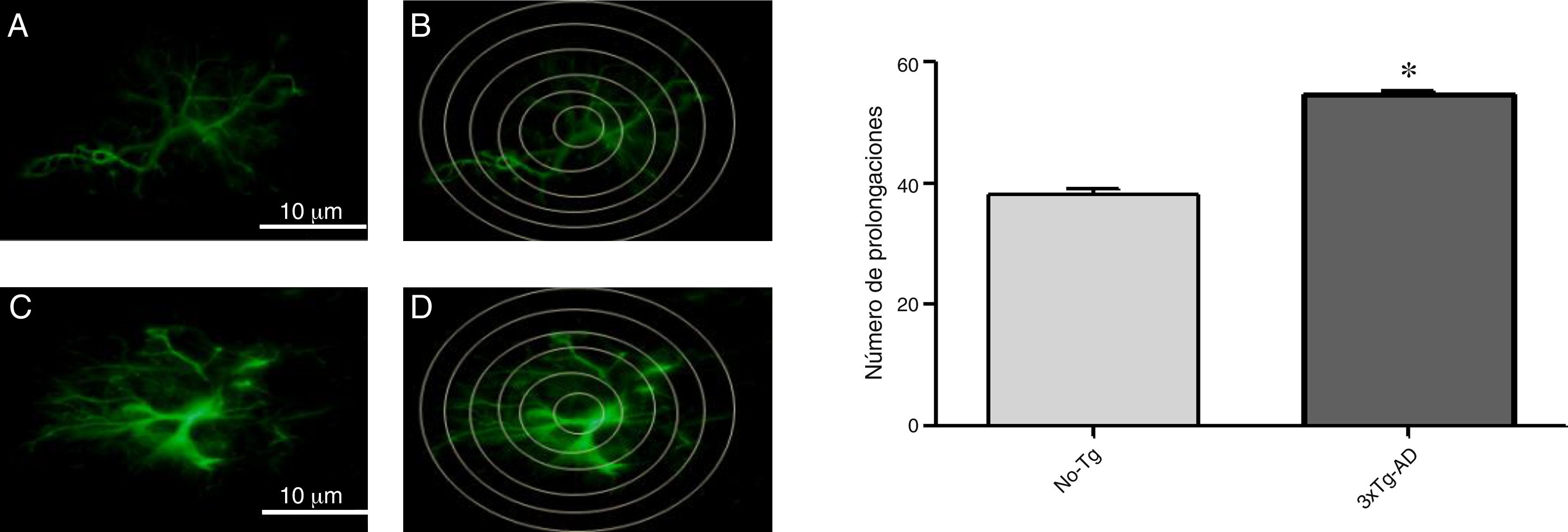
En el análisis de la inmunorreactividad de GFAP se observó aumento en la M1 del ratón 3xTg-AD, al compararlo con el ratón No-Tg. Los astrocitos positivos a GFAP mostraron signos de reactividad, hipertrofia, alargamiento de cuerpos celulares y aumento en el volumen y superficie. En la figura 3 se muestra el aumento de las intersecciones de las prolongaciones de los astrocitos inmunorreactivos para GFAP (círculo de Sholl, 1953 modificado), del 3xTg-AD al compararlo con el No-Tg.
. B y D) Las intersecciones inmunorreactivas de GFAP en donde se sobrepone el círculo de Sholl y se tomaron en cuenta para el análisis en esta región cerebral en ratón no-transgénico (B) y en el ratón triple transgénico (D), respectivamente. Las gráficas indican el promedio±EEM de 6 experimentos, con el aumento significativo (*p<0,05) del número de las prolongaciones inmunorreactivas a GFAP medidas en las intersecciones de los círculos concéntricos en el 3xTg-AD al compararlo con el No-Tg.")
Fotomicrografías de la inmunorreactividad de GFAP en la M1 de ratones hembra de 11 meses (A y C, No-Tg y 3xTg-AD, respectivamente). B y D) Las intersecciones inmunorreactivas de GFAP en donde se sobrepone el círculo de Sholl y se tomaron en cuenta para el análisis en esta región cerebral en ratón no-transgénico (B) y en el ratón triple transgénico (D), respectivamente. Las gráficas indican el promedio±EEM de 6 experimentos, con el aumento significativo (*p<0,05) del número de las prolongaciones inmunorreactivas a GFAP medidas en las intersecciones de los círculos concéntricos en el 3xTg-AD al compararlo con el No-Tg.
Los factores de riesgo que propician la EA de tipo esporádico están ligados al comportamiento humano desde, los hábitos en la dieta, el consumo de tabaco, hasta la contaminación de tipo ambiental29. Los estudios epidemiológicos indican que personas con bajos niveles de educación, con un historial de trauma cerebral, con consumo alto de calorías, o un estilo de vida sedentaria, son las que tienen el mayor riesgo de presentar la enfermedad30. En el cerebro humano las alteraciones en las áreas isocorticales de asociación de alto orden multimodal son responsables del deterioro progresivo de las capacidades cognitivas, que incluyen la disfunción ejecutiva (corteza prefrontal) y memoria semántica2,13,15.
Es claro que las alteraciones físicas y funcionales en la EA progresan conforme avanza la patología y, aunque en menor grado, el aparato locomotor también se compromete3. Así, se registran las alteraciones motoras durante la primera etapa de la EA como apatía y tendencia al sedentarismo que favorece la inmovilización, y acelera el deterioro físico31. En la segunda etapa (intermedia), se inician las alteraciones del control de la postura y de la marcha, lo que requiere la actividad física se asocia con bajo riesgo de disfunción cognitiva en la EA, en demencias en general favoreciendo la memoria32. Está claro que las regiones sensoriales y motoras del sistema nervioso central están afectadas por la patología de Alzheimer y que las intervenciones dirigidas a mejorar los déficits sensorio-motores pueden mejorar la función del paciente a medida que avanza la patología33. En este sentido, varios experimentos se llevan a cabo con fisioterapia y/o ejercicio voluntario en modelos transgénicos para la EA, en donde se demuestran que el ejercicio y/o el enriquecimiento del medio ambiente puede ayudar a la supervivencia neuronal y la resistencia a insulto cerebral, promover la vascularización cerebral, estimular la neurogénesis, mejorar el aprendizaje y contribuir al mantenimiento de la función cognitiva34. En el caso del ratón 3xTg-AD, se ha demostrado que el efecto de la rueda de ejercicio representa una manipulación ambiental y provoca plasticidad neuro comportamental en términos de interacciones gen-ambiente relevantes para la patogénesis34.
En el diseño de este estudio, se encontró una relación con el deterioro en la prueba motora de campo abierto (conducta en la que se involucra la corteza motora) con las alteraciones en las neuronas, la amiloidogénesis y astrogénesis en esta región cortical del 3xTg-AD en etapa intermedia de la (11 meses de edad). En la EA, una de las principales alteraciones neuropatológicas descritas es la disminución del peso y volumen cerebral, que se asocia a la disminución neuronal35. Esta observación se relaciona con el presente estudio, en donde se observó una alteración neuronal significativa en el modelo 3xTg-AD en comparación el grupo No-Tg, lo que sugiere que en este modelo las células neuronales en la M1 son susceptibles a daño neuronal (hipercromatismo citoplasmático, encogimiento celular y fragmentación nuclear). Estos hallazgos se les puede comparar con aquellos en donde se utilizan modelos de ratón transgénico para la EA y en donde se reportó pérdida neuronal pero además de la corteza, en el hipocampo, estructura relacionada con la memoria de tipo espacial15,35,36. El mecanismo propuesto es la sinaptopatía, en donde se dañan los circuitos corticales que no mantienen los contactos sinápticos en las redes celulares que participan en funciones cognitivas en el adulto24. En este estudio, también se encontró la presencia de los depósitos extracelulares de βA y de la tau hiperfosforilada en la corteza como ha sido reportado17,36 y asociado a la deficiencia cognitiva, confirmándolo como modelo que presenta la patología de la forma humana y que la deficiencia en la prueba del campo abierto lo potencia como indicativo también del deterioro motor asociado a la presencia de las características patológicas de ambas proteínas (βA y tau), pareciendo que la proteína tau sea la principal responsable en la muerte de las neuronas en la EA. Además, se ha reportado que la actividad celular del hipocampo, para la ubicación del lugar puede contribuir a las representaciones espaciales inestables y los déficits de la memoria espacial37.
Por otra parte, la forma monomérica o soluble del péptido βA es tóxico para las neuronas y se ha demostrado por varias observaciones in situ e in vitro que desencadena la reacción inflamatoria en el cerebro de pacientes con EA38. Específicamente, la microglía produce una serie de citocinas proinflamatorias y mediadores en respuesta a βA y activa a los astrocitos, que se convierten en parte del proceso inflamatorio, creando un círculo vicioso neuroinflamatorio39. Por otra parte, se estudian las vías que regulan la inmunidad innata en el cerebro relacionada con este péptido para su posible intervención terapéutica40. En el cerebro, las placas βA están rodeadas por astrocitos reactivos, como un mecanismo defensivo endógeno contra la deposición de la placa, pero la activación persistente y la inflamación asociada también pueden contribuir a la progresión de la EA41. Los astrocitos se comunican activamente con las neuronas, modulando así la plasticidad sináptica a través de la liberación de gliotransmisores, como el glutamato y la D-serina39, y en la EA, los astrocitos pueden sufrir cambios morfológicos y funcionales, denominados astrogliosis, evidenciada en este estudio por el aumento de la expresión de la proteína ácida fibrilar glial22,42 y además por la hipertrofia astrocitaria reportada21,26, y por las alteraciones anatómicas valoradas como aumento en sus prolongaciones y que pudiera estar asociada con el aumento de la producción de factores que pueden ser dañinos para las células circundantes41. Por lo tanto, los procesos neurodegenerativos de los astrocitos repercutirán en la defensa frente al daño, al activar una amplia variedad de funciones neuroprotectoras42,43.
En tejido post mortem de pacientes con la EA también se ha reportado astrogliosis en comparación con los casos control, e incluso e esta aumenta en función de la edad44. Por lo tanto, los astrocitos son elementos clave en el desarrollo de la demencia tipo Alzheimer45. Nuestros resultados son consistentes con el aumento de la inmunorreactividad para GFAP en el hipocampo al comparar con la corteza cerebral motora primaria. Pudiera entonces, ocurrir que esta hipertrofia astrocítica va en aumento conforme avanza la patología y la microglía, así como las células inmunes influyen en el inicio de la enfermedad, en la progresión o en ambas etapas. Incluso el péptido βA que se agrega extracelularmente en las placas neuríticas típicas generan un entorno inflamatorio constante potenciando el daño neuronal y alteración de la barrera hematoencefálica45.
Así, la disfunción neuronal y glial y las vías que regulan la inflamación en el cerebro tienen un papel importante en las etapas intermedias de la enfermedad, en donde los mecanismos plásticos neuronales están limitados y el daño es irreversible. Por lo que es importante determinar las etapas previas y establecer estrategias para su posible intervención terapéutica en los periodos vulnerables para detener los procesos degenerativos que conllevan a la enfermedad.
FinanciaciónEste trabajo fue parcialmente financiado por el CONACYT (no. 295523; Proyecto No. CB-2012/178841) y UNAM-DGAPA (IN203616).
Conflicto de interesesLos autores no tienen conflictos de interés para declarar.
Los autores desean agradecer a las Unidades de Microscopia, de Proteogenómica y de conducta (Dra. Deysi Gasca) del INB y a los técnicos de laboratorio A. Aguilar Vázquez y M. Servín García, por su ayuda en el cuidado de los animales.

