



El síndrome de Struge-Weber (SSW) o angiomatosis encefalotrigeminal es una neuroectodermatosis, de presentación esporádica, caracterizado por: angioma facial en territorio trigeminal, angioma leptomeníngeo ipsilateral y angioma coroideo ipsilateral. Se han descrito variantes clínicas con diferentes combinaciones de esta tríada. El diagnóstico se hace fundamentalmente en la infancia, pero en ocasiones excepcionales se diagnostica en la edad adulta. Presentamos un caso con angiomatosis leptomeníngea aislada y de inicio de síntomas en la edad adulta.
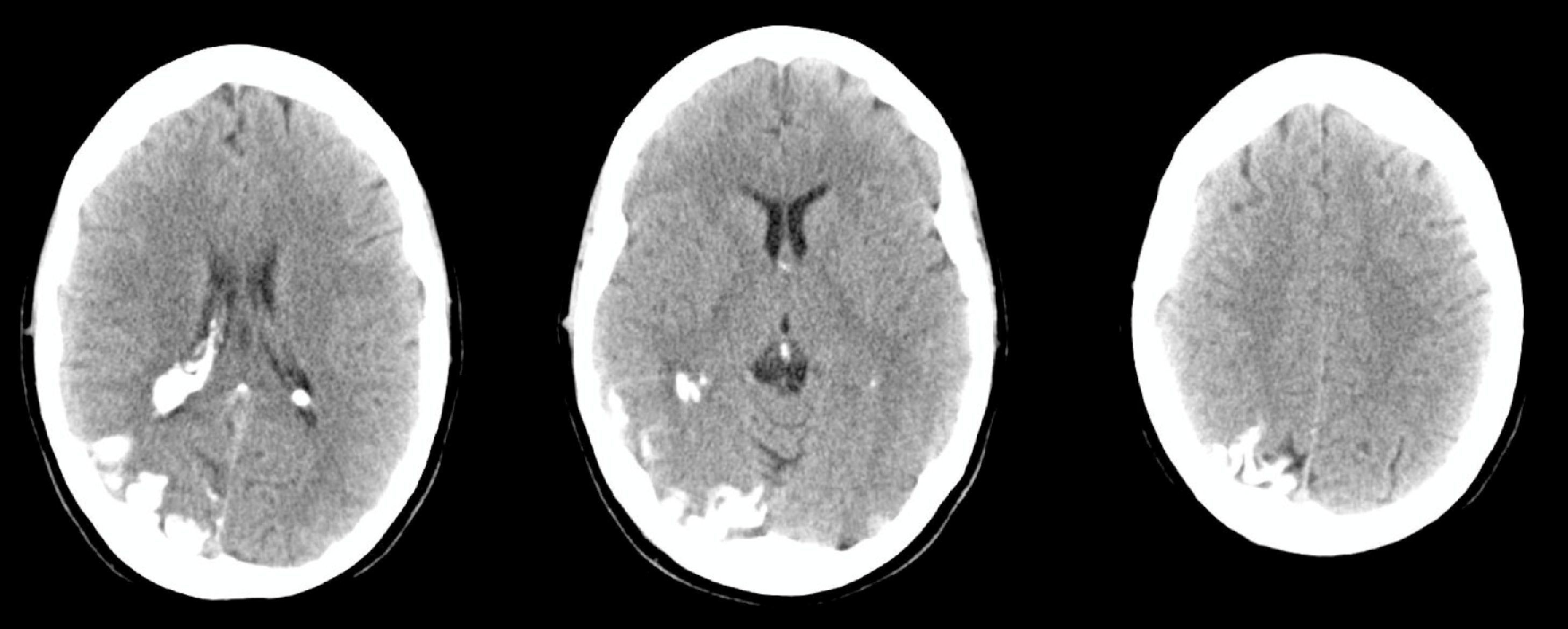
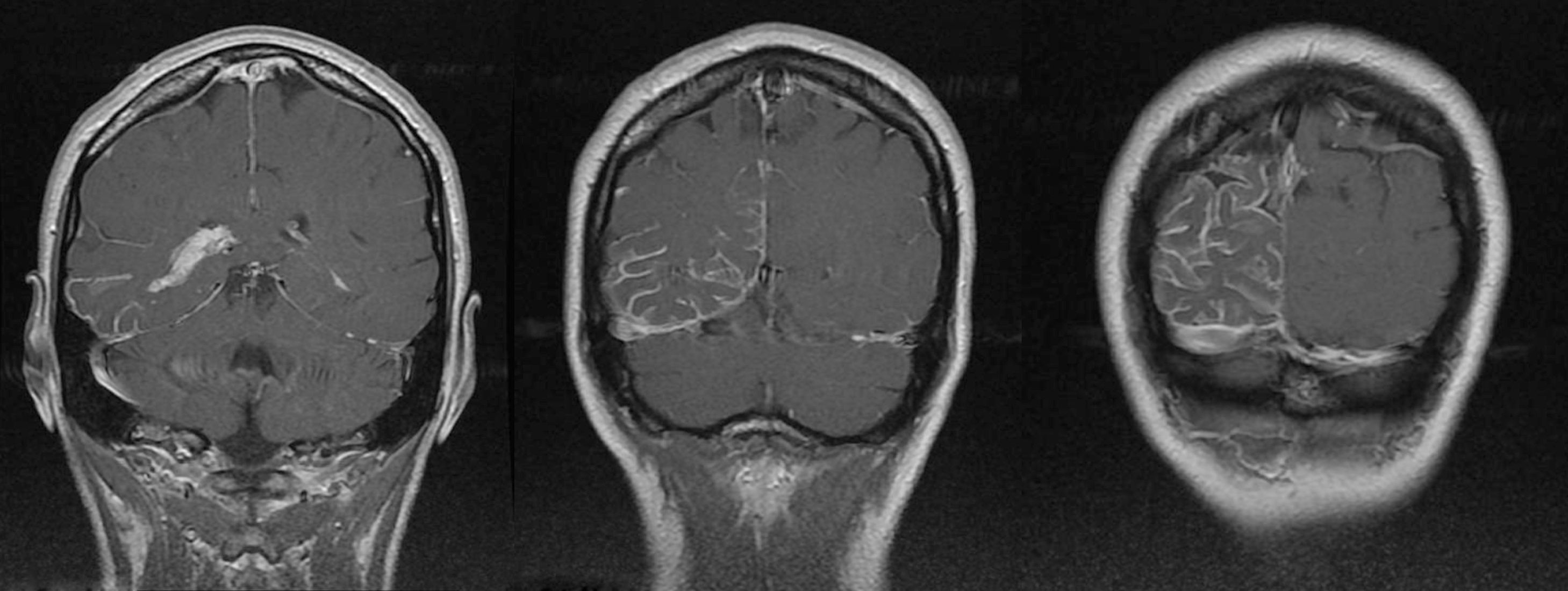
Se trata de una mujer de 44 años, sin antecedentes de interés, que acude a Urgencias por cuadro de cefalea súbita, hemicraneal derecha, intensa, opresiva, con escasa respuesta a analgésicos, que a las 24 h asocia desorientación, agitación psicomotriz y alucinaciones visuales. A la exploración física presenta lesión herpética en el labio superior. En la analítica destaca leucocitosis con desviación izquierda y proteína C reactiva (PCR) elevada, en una tomografía axial computarizada (TAC) craneal sin y con contraste (fig. 1) se objetiva hiperdensidad giriforme parieto-occipital derecha y realce meníngeo focal, informado inicialmente como compatible con meningitis crónica. Se realiza una punción lumbar que muestra acelularidad con hiperproteinorraquia y motiva la introducción transitoria de vancomicina, ceftriaxona y aciclovir iv por el personal del servicio de urgencias ante la posibilidad de meningoencefalitis. Ingresa en planta de Neurología, donde permanece afebril, con cefalea intensa acompañada de náuseas, vómitos y episodios fluctuantes de alucinaciones visuales y auditivas. La exploración neurológica demuestra una hemianopsia homónima izquierda. Se realiza una resonancia magnética nuclear (RMN) craneal con y sin gadolinio (fig. 2), donde se objetiva una angiomatosis leptomeníngea en el hemisferio cerebral derecho (parietal-occipito-temporal). En el electroencefalograma, la amplitud de la actividad de base se encuentra atenuada sobre región temporo-occipital derecha. Ante la ausencia de datos de infección (y 2 hemocultivos, PCR y cultivos de LCR negativos) y una segunda punción lumbar acelular pero con presión de apertura elevada e hiperproteinorraquia, se retira la antibioterapia empírica. Una interconsulta con Oftalomogía descarta la presencia de malformación vascular ocular. Se diagnostica a la paciente de síndrome de Sturge-Weber sin angioma facial, con crisis parciales temporo-occipitales y posible trombosis/estasis venoso en seno de angiomatosis, y se instaura tratamiento con acetazolamida (por la hipertensión intracraneal), levetiracetam y ácido acetilsalicílico, con resolución de la cefalea y los episodios confuso-alucinatorios, cuya naturaleza pensamos que tenga un origen comicial y/o fenómeno vascular seudomigrañoso. Se ha realizado el seguimiento clínico de la paciente en consultas externas de Neurología, tras una nueva punción lumbar, acelular con normoproteinorraquia y presión de apertura normal, se ha descendido paulatinamente la dosis de acetalozamida hasta su supresión, manteniéndose la paciente asintomática tras un año de seguimiento.

 en los surcos cerebrales. Asimetría en el tamaño entre ambos plexos coroideos en los astas temporales de los ventrículos laterales, siendo llamativamente de mayor tamaño y con mayor realce en el ventrículo lateral derecho.")
RMN craneal: cortes axiales de imágenes potenciadas en T1 con contraste; llamativo realce meníngeo de distribución predominantemente parietal-occipito-temporal, siendo el realce tanto dural como, y sobre todo, leptomeníngeo (pial) en los surcos cerebrales. Asimetría en el tamaño entre ambos plexos coroideos en los astas temporales de los ventrículos laterales, siendo llamativamente de mayor tamaño y con mayor realce en el ventrículo lateral derecho.
La angiomatosis encefalotrigeminal o SSW es un síndrome neurocutáneo poco frecuente, de diagnóstico generalmente en la infancia, con presentación esporádica, que afecta a la microvascularización venosa cefálica y que, clínicamente, en su forma típica se caracteriza por angioma facial plano de color rojo vinoso en territorio de inervación de la rama oftálmica del nervio trigémino, situado ipsilateralmente a una angiomatosis leptomeníngea a nivel del lóbulo occipital y parietal, junto con malformación vascular ocular. Existen variantes incompletas1 del síndrome que aparecen como: a) angioma facial y leptomeníngeo, sin angioma coroideo; b) angioma leptomeníngeo y coroideo sin nevus facial; c) nevus facial y angioma coroideo sin evidencias clínicas ni radiográficas de angiomatosis cerebral, y d) angiomatosis cerebral y pial aislada2-4.
Aunque la descripción del SSW se remonta a 1879 (Sturge), en el año 1935, Van Bogaert describe una forma de angiomatosis córtico-meníngea-telangiectásica sin angioma facial como variante del SSW. Lund, en 1949, en una revisión de 144 pacientes de la literatura afectados del síndrome, halla 7 con imágenes radiográficas típicas y sin angioma facial5. Existen pocas comunicaciones de casos de SSW sin angioma facial en la literatura médica6-9 y el diagnóstico de estos en la edad adulta, como en nuestro caso, es excepcional10.
Las manifestaciones neurológicas del SSW se relacionan con la presencia del angioma leptomeníngeo11. Lo más constante es la presencia de crisis epilépticas que afectan al 75-90% de los pacientes2 y solo el 7% inicia las crisis después de los 5 años. Los déficits neurológicos pueden aparecer lentamente en el tiempo, o bien como episodios «stroke-like» asociados a crisis y/o cefalea de rasgos migrañosos como en el caso clínico descrito. Los signos de déficit neurológico que se muestran con mayor frecuencia son: déficit motor hemiparético, hemianopsia y hemiatrofia corporal, y se observan en el 65% de los enfermos. Las alteraciones cognitivas son frecuentes y se relacionan con el inicio precoz de crisis de difícil control y afectación cerebral bilateral12. Los síntomas depresivos son también comunes. Las cefaleas y las migrañas aparecen con frecuencia, y se incluyen en la segunda clasificación de la Internacional Headache Society (IHS), dentro de las cefaleas secundarias, en el apartado 6.313.
El diagnóstico de la afectación cerebral se realiza, ante la sospecha clínica, mediante técnicas de neuroimagen, de gran utilidad en ausencia de las alteraciones cutáneas u oculares típicas. La característica radiográfica de esta enfermedad son las calcificaciones corticales giriformes adyacentes al angioma leptomeníngeo, que aparencen en la TAC craneal. En la RMN craneal, se visualiza la malformación vascular típica, que realza con contraste iv en imágenes potenciadas en T1. Asociado a esto, se puede encontrar dilatación y realce del plexo coroideo ipsilateral y dilatación del drenaje venoso profundo subyacente a la región cortical afectada.
El tratamiento con fármacos antiepilépticos de manera prolongada es fundamental en aquellos pacientes que presentan crisis comiciales. Puesto que el estasis venoso y la trombosis microvascular contribuyen probablemente al deterioro neurológico en el SSW, se recomienda empíricamente antiagregación diaria con dosis bajas de ácido acetilsalicílico14, demostrando una disminución de la frecuencia de crisis comiciales y eventos «stroke-like». Las cefaleas y las migrañas se tratan de la manera habitual, con medicación abortiva (incluso triptanes15) y, si es preciso, por la frecuencia o la severidad de los episodios, con fármacos preventivos. La cirugía (resección quirúrgica de la malformación e incluso hemisferectomía) es efectiva, pero no debe ser considerada en el momento del diagnóstico, ya que la indicación actual es la presencia de crisis refractarias al tratamiento médico.
A la Dra. Noelia Arévalo Galeano, de la Unidad Central de Radiodiagnóstico, por su ayuda en este caso.

