




El síndrome X frágil (SXF) es la causa más frecuente de discapacidad intelectual hereditaria y se asocia a un amplio espectro de enfermedades en las distintas generaciones de una misma familia. En este trabajo se revisan las manifestaciones clínicas de los trastornos asociados al X frágil y el espectro de mutaciones en el gen 1 del retraso mental del X frágil (FMR1), la neurobiología de la proteína del retardo mental X frágil (FMRP) y una visión general de los potenciales blancos terapéuticos y el asesoramiento genético.
DesarrolloEsta enfermedad es causada por una amplificación de las repeticiones CGG (>200 repeticiones) en la región 5’ no traducida del gen FMR1, que lleva al déficit o ausencia de la proteína FMRP. La FMRP es una proteína de unión al ARN que regula la traducción de varios genes que son importantes en la plasticidad sináptica y la maduración dendrítica. Se cree que expansiones de las repeticiones CGG en el rango de premutación (55-200 repeticiones) generan un aumento en los niveles de mRNA de FMR1, lo que produciría toxicidad neuronal. Esto se manifiesta en problemas del desarrollo tales como autismo y problemas de aprendizaje, así como en patologías neurodegenerativas como el síndrome de temblor/ataxia asociado al X frágil (FXTAS).
ConclusionesLos avances en la identificación de las bases moleculares del SXF pueden servir como modelo para comprender las causas de las enfermedades neuropsiquiátricas y probablemente conducirán al desarrollo de tratamientos cada vez más específicos.
Fragile X syndrome, the most common inherited cause of intellectual disability, is associated with a broad spectrum of disorders across different generations of a single family. This study reviews the clinical manifestations of fragile X-associated disorders as well as the spectrum of mutations of the fragile X mental retardation 1 gene (FMR1) and the neurobiology of the fragile X mental retardation protein (FMRP), and also provides an overview of the potential therapeutic targets and genetic counselling.
DevelopmentThis disorder is caused by expansion of the CGG repeat (>200 repeats) in the 5 prime untranslated region of FMR1, resulting in a deficit or absence of FMRP. FMRP is an RNA-binding protein that regulates the translation of several genes that are important in synaptic plasticity and dendritic maturation. It is believed that CGG repeat expansions in the premutation range (55 to 200 repeats) elicit an increase in mRNA levels of FMR1, which may cause neuronal toxicity. These changes manifest clinically as developmental problems such as autism and learning disabilities as well as neurodegenerative diseases including fragile X-associated tremor/ataxia syndrome (FXTAS).
ConclusionsAdvances in identifying the molecular basis of fragile X syndrome may help us understand the causes of neuropsychiatric disorders, and they will probably contribute to development of new and specific treatments.
Se estima que entre 1-3% de la población mundial presenta discapacidad intelectual (DI)1, siendo el síndrome de X frágil (SXF) la causa más frecuente de DI hereditaria en varones2 y la principal enfermedad monogénica asociada a autismo3. El SXF afecta a uno de cada 4.000 varones y a una de cada 8.000 mujeres2, pero podría ser más frecuente si se consideran los trastornos de conducta y DI leves4. Casi la mitad de los casos de DI ligada al cromosoma X corresponden a SFX5.
El SXF presenta una herencia dominante ligada al cromosoma X, con penetrancia incompleta y forma parte de un grupo de enfermedades asociadas a mutaciones del gen 1 del retardo mental X frágil (FMR1), denominadas trastornos asociados al X frágil (TASXF), entre los cuales se encuentran el síndrome de temblor/ataxia asociado al X frágil (FXTAS) y el síndrome de insuficiencia ovárica primaria asociada al X frágil (FXPOI)6.
El SXF se caracteriza clínicamente por DI y rasgos físicos tales como orejas grandes y prominentes, hiperlaxitud de las articulaciones y pie plano7. En varones se describe estrabismo, facie alargada con mentón prominente, pectum excavatum, prolapso de la válvula mitral y macroorquidismo después de los 8 años8,9. Sin embargo, un 30% de los casos no presentan los rasgos fenotípicos clásicos ni historia familiar de DI, manifestándose como retraso del lenguaje o trastorno de déficit atencional con/sin hiperactividad (TDAH), lo que dificulta y retrasa el diagnóstico7.
El compromiso cognitivo se manifiesta precozmente, con retraso del desarrollo psicomotor10, movimientos repetitivos, posturas inusuales, pobre contacto ocular y aislamiento social11. En los niños que tienen rasgos autistas (30%), el compromiso cognitivo, el retraso del lenguaje y los trastornos adaptativos son más severos12. En algunos casos el retraso psicomotor puede ser leve o incluso ser inicialmente normal, manifestándose posteriormente como trastorno de aprendizaje13. El 85% de los hombres y un 25-30% de las mujeres tienen un coeficiente intelectual menor de 70; entre las mujeres se observa con mayor frecuencia una inteligencia normal o limítrofe14. La mayoría de los pacientes logra desarrollar un lenguaje oral, conocimientos generales y destrezas en las actividades de la vida diaria15,16.
Por otra parte, un 13-18% de los varones y un 4% de las mujeres presentan epilepsia, con convulsiones generalizadas o parciales complejas17,18. La epilepsia es 3 veces más frecuente en pacientes con SXF que presentan rasgos autistas17.
Los pacientes manifiestan además importantes trastornos emocionales, suelen ser ansiosos, hiperactivos y en algunos casos son irritables, inflexibles y muestran agresividad19,20. En las mujeres son más frecuentes los problemas emocionales, incluyendo depresión, ansiedad y retraimiento21.
Algunos pacientes pueden tener un fenotipo similar al síndrome de Prader Willi, con DI, obesidad, hiperfagia y retraso de la pubertad, que puede asociarse a autismo, pero no presentan hipogonadismo22.
Mutaciones en el gen 1 del retraso mental del X frágil (FMR1) y trastornos asociadosEn más del 98% de los casos el SXF es causado por una mutación completa (MC), que es producida por la amplificación del triplete citosina-guanina-guanina (CGG) (>200 repeticiones) en la región 5′ no traducida del gen FMR1, locus FRAXA ubicando en Xq27.323.
Los individuos sanos tienen entre 4-45 repeticiones CGG en esta región, lo cual permite la estabilidad durante la replicación del ADN. Amplificaciones de más de 50 repeticiones confieren inestabilidad a la región, pudiendo transformarse en una mutación completa en las siguientes generaciones24.
La MC produce una hipermetilación de la isla de citosina-fosfato-guanina (CpG) ubicada en la región promotora del gen FMR1, lo que provoca un cambio conformacional de la cromatina, volviéndose más condensada. Dicha condensación produce la inhibición completa o casi total de la transcripción del gen, niveles escasos o nulos de su ácido ribonucleico mensajero (mRNA) y una reducción significativa de los niveles de su producto o su completa ausencia, la proteína del retardo mental X frágil (FMRP)25,26. Sin embargo, también se ha visto últimamente que el silenciamiento del gen FMR1 se produciría por su propio mRNA expandido a través de la hibridación entre las porciones complementarias de repeticiones CGG, formando un complejo RNA-DNA e impidiendo su expresión27.
Con menor frecuencia, el SXF puede ocurrir por una mutación puntual, deleción del gen FMR1 o su promotor28, o por una expansión menor de las repeticiones CGG (premutación [PM]) que es capaz de producir bajos niveles de FMRP y DI29,30.
Las diferencias fenotípicas observadas entre mujeres con MC, y entre hombres y mujeres con MC, se explican por los distintos niveles de expresión de FMRP y por una inactivación no al azar del cromosoma X mutado. Las mujeres con inactivación preferencial del cromosoma X normal presentan mayor DI y niveles bajos de FMRP31,32, presentando un fenotipo físico y neuropsiquiátrico similar al observado en varones, pero más leve.
Una amplificación del triplete CGG entre 55-200 repeticiones se denomina PM, condición que se asocia a inestabilidad replicativa, especialmente durante la gametogénesis femenina. En la población general se presenta en una de 113-259 mujeres y en uno de 260-813 varones2. La PM no afecta sustancialmente la expresión de la proteína FMRP, sin embargo, expansiones sobre 100 repeticiones CGG frecuentemente llevan a una MC y SXF en la próxima generación29.
En portadores de PM se describe leve compromiso cognitivo y problemas de conducta, especialmente en varones33. El compromiso clínico está relacionado con la toxicidad producida por los niveles elevados de mRNA del gen FMR18,34. La PM también se asocia a insuficiencia ovárica primaria (FXPOI, 20% de las mujeres portadoras), manifestándose con el cese de menstruaciones antes de los 40 años, y a síndrome de temblor/ataxia asociado a X frágil (FXTAS) en varones (40%) y mujeres (8-16%) en edad adulta35,36. El FXTAS tiene criterios diagnósticos bien establecidos, y aunque se están cuestionando actualmente, son de gran utilidad para establecer un diagnóstico de definitivo, probable o posible de FXTAS37 (tabla 1). Dado que un 2-4% de los hombres con ataxia cerebelar de inicio tardío (después de los 50 años) podrían ser portadores de una PM, se recomienda estudiar el número de repeticiones CGG en FMR1 en todas las personas mayores de esta edad con este tipo de sintomatología38,39. Hombres y mujeres portadoras de PM tienen además mayor frecuencia de enfermedades autoinmunes, incluyendo hipotiroidismo y fibromialgia37.
Criterios diagnósticos y categorías diagnósticas de FXTASa
| Criterios mayores | Criterios menores | |
|---|---|---|
| Molecular 55-200 repeticiones Radiológicos (a la RM*) Lesiones hiperintensas en secuencias T2 en sustancia blanca de pedúnculos cerebelosos medios Clínicos Temblor de intención Marcha atáxica Neuropatológicos Inclusiones FXTAS | Radiológicos (a la RM)b Lesiones hiperintensas en secuencias T2 en sustancia blanca cerebral Atrofia generalizada moderada a severa Clínicos Parkinsonismo Déficit moderado a severo de la memoria de corto plazo Déficit de funciones ejecutivas | |
| Categorías diagnósticas: requieren de la presencia del criterio molecular | ||
| Definitivo Presencia de un signo radiológico mayor más (i) un criterio clínico mayor o (ii) la presencia de inclusiones FXTAS | Probable Presencia de (i) un criterio radiológico mayor y un criterio clínico menor o (ii) 2 criterios clínicos mayores | Posible Presencia de un criterio radiológico menor y un criterio clínico mayor |
La expansión de las repeticiones CGG es mitóticamente inestable favoreciéndose la heterogeneidad somática y la presencia de mutaciones en mosaico. Un individuo mosaico para el tamaño del alelo tiene alelos con MC y alelos con PM (12% de los pacientes afectados con SXF)5,30. También es posible que ocurra un mosaico de metilación, término que se refiere a la presencia de alelos con MC hipermetilados y alelos desmetilados amplificados en el rango de PM y/o MC (6% de los pacientes). En los casos de mutación en mosaico hay cantidades variables de FMRP y un mayor coeficiente intelectual14,30.
Otro punto a destacar es que las repeticiones CGG pueden verse interrumpidas por tripletes AGG, cuya cantidad y posición son importantes en la estabilidad replicativa y por ende, en la posibilidad de expansión en la siguiente generación. De hecho, se ha establecido que el riesgo de expansión a MC en hijos de madres portadoras de una PM disminuye en un 60% si la madre presenta 2 interrupciones AGG dentro de una repetición total de entre 70-80 tripletes CGG, en comparación con aquellas madres que no tienen ninguna interrupción dentro del mismo rango40.
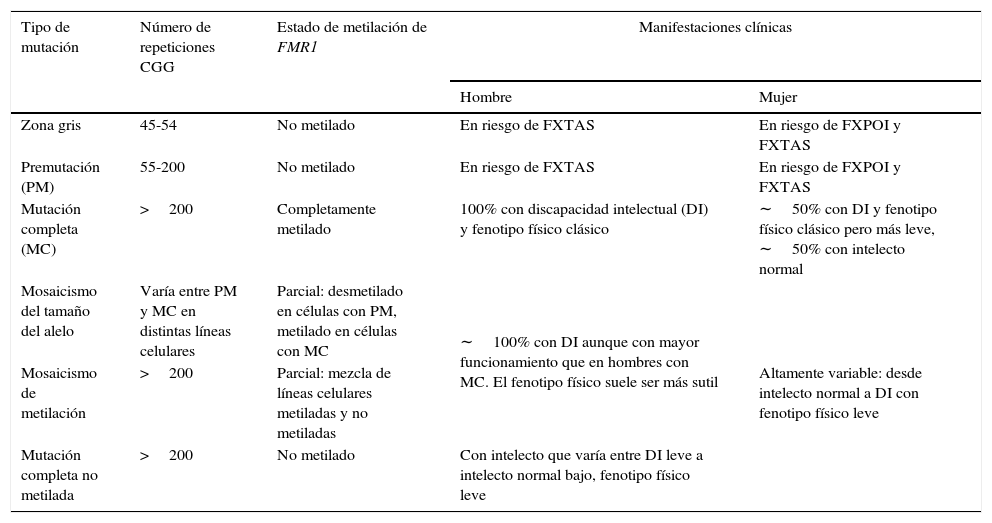
Por último, expansiones entre 45-54 CGG corresponden a la zona intermedia o zona gris que intercala a alelos entre los rangos normales y PM. Las expansiones de la zona gris no están claramente asociadas a un fenotipo determinado, sin embargo, pueden llevar a una MC al cabo de 2 generaciones y las personas portadoras tendrían un riesgo levemente incrementado de presentar FXTAS y/o FXPOI37,41. De esta manera, el amplio espectro clínico de las mutaciones del gen FMR1 y los desórdenes asociados hacen que las familias presenten manifestaciones genotípicas y fenotípicas diversas a través de las generaciones42,43 (fig. 1 y tabla 2).
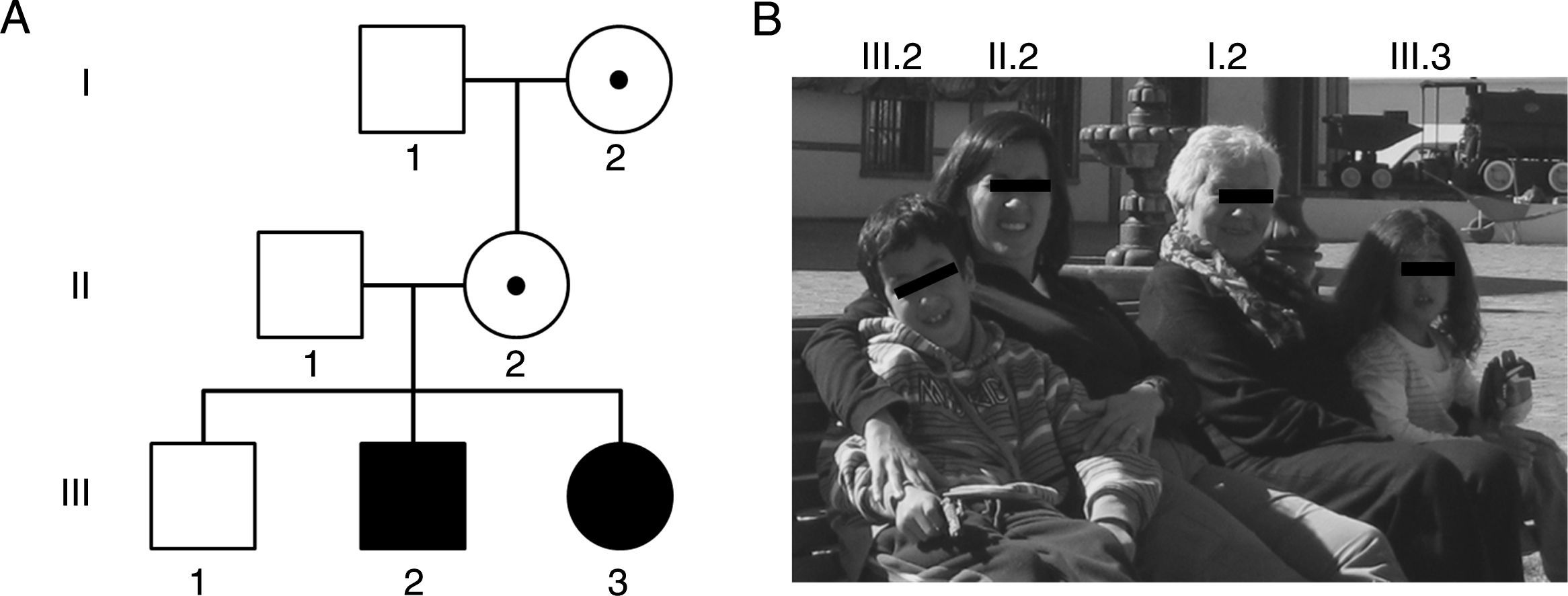
 En la genealogía, los individuos I.2 y II.2 presentan una premutación de 83 y 96 tripletes CGG y manifestaron insuficiencia ovárica prematura a los 35 y 36 años, respectivamente. Los individuos III.2 y III.3 son hermanos que presentan una mutación completa de 370 y 570 tripletes CGG, y fueron diagnosticados con SXF a los 5 y un año, respectivamente. B) Fotos de los integrantes de la familia descrita y su respectiva ubicación en la genealogía.")
Familia y su genealogía con espectro de desórdenes relacionados con el SXF. A) En la genealogía, los individuos I.2 y II.2 presentan una premutación de 83 y 96 tripletes CGG y manifestaron insuficiencia ovárica prematura a los 35 y 36 años, respectivamente. Los individuos III.2 y III.3 son hermanos que presentan una mutación completa de 370 y 570 tripletes CGG, y fueron diagnosticados con SXF a los 5 y un año, respectivamente. B) Fotos de los integrantes de la familia descrita y su respectiva ubicación en la genealogía.
Tipos de mutaciones en FMR1 y manifestaciones clínicas asociadasa
| Tipo de mutación | Número de repeticiones CGG | Estado de metilación de FMR1 | Manifestaciones clínicas | |
|---|---|---|---|---|
| Hombre | Mujer | |||
| Zona gris | 45-54 | No metilado | En riesgo de FXTAS | En riesgo de FXPOI y FXTAS |
| Premutación (PM) | 55-200 | No metilado | En riesgo de FXTAS | En riesgo de FXPOI y FXTAS |
| Mutación completa (MC) | >200 | Completamente metilado | 100% con discapacidad intelectual (DI) y fenotipo físico clásico | ∼50% con DI y fenotipo físico clásico pero más leve, ∼50% con intelecto normal |
| Mosaicismo del tamaño del alelo | Varía entre PM y MC en distintas líneas celulares | Parcial: desmetilado en células con PM, metilado en células con MC | ∼100% con DI aunque con mayor funcionamiento que en hombres con MC. El fenotipo físico suele ser más sutil | Altamente variable: desde intelecto normal a DI con fenotipo físico leve |
| Mosaicismo de metilación | >200 | Parcial: mezcla de líneas celulares metiladas y no metiladas | ||
| Mutación completa no metilada | >200 | No metilado | Con intelecto que varía entre DI leve a intelecto normal bajo, fenotipo físico leve | |
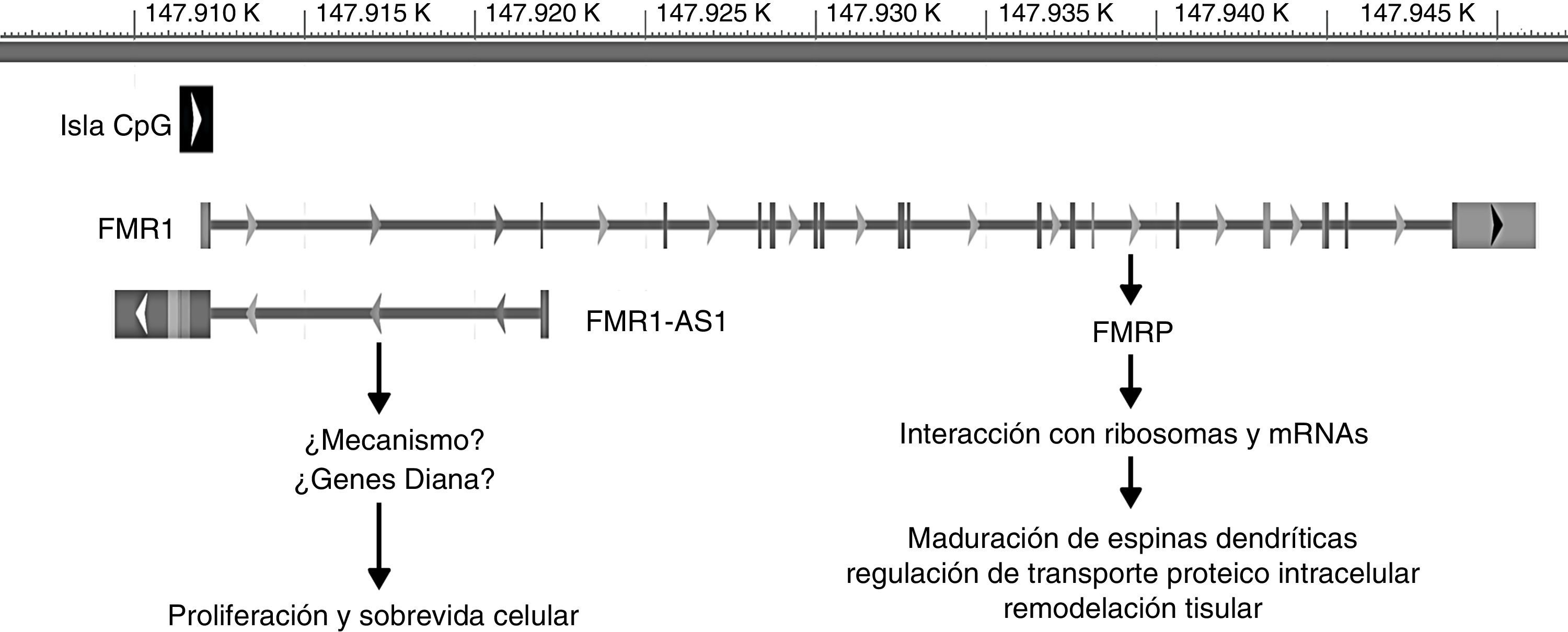
Se ha visto en el último tiempo que el gen FMR1 ejerce su función a través de 2 mecanismos: el más conocido está en relación con su producto proteico FMRP y el otro en relación con su otro producto, el RNA largo no codificante (lncRNA) FMR4 o FMR1-AS1. Los lncRNA son genes que codifican RNA que participan en la regulación transcripcional o traduccional de otros genes, tanto de manera positiva como negativa44,45. El promotor del gen FMR1 codifica en la orientación antisentido el gen FMR4, el cual se superpone a la región de repeticiones CGG y la isla CpG. De la misma forma que FMR1, FMR4 está silenciado en pacientes con MC y está sobreexpresado en pacientes portadores de PM45 (fig. 2).
![Ubicación genómica y rol de FMR1 y FMR1-AS1. La barra gris representa la posición genómica (en kilobases [K]) de ambos genes en el cromosoma X desde el extremo p-terminal. La caja negra simboliza la isla CpG involucrada en el silenciamiento de ambos genes en la mutación completa. Las flechas ubicadas en esta isla así como en cada gen simbolizan la orientación de la transcripción. Las líneas verticales en FMR1 representan los exones de este gen.](https://static.elsevier.es/multimedia/02134853/0000003200000004/v1_201704150014/S0213485314002321/v1_201704150014/es/main.assets/gr2.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNcnxJVYM2BEAEuZ5XZLhi9i6WSMfelRHtJaOSE9bGYOuZpAEskOvZ38NrvZTgRjNkoTsjvtRBjaFr2r8U2wpzf0a1sLH0JC6Isv2/PvlVoy+U8gg07q+nY2nw20IOqVvmKaM/IAO2lrVtbOMhcxwwhyIvsMMxp+M6cKlu25srlqLMA4fX4gRFUy3VdBxzYTUltR7E4eYPtGNDXlRnZK1XdmqBrTuY8l2XzU4Gl0vBQ+2VpedTfOFqdqUnLXRxObiQQ= "Ubicación genómica y rol de FMR1 y FMR1-AS1. La barra gris representa la posición genómica (en kilobases [K]) de ambos genes en el cromosoma X desde el extremo p-terminal. La caja negra simboliza la isla CpG involucrada en el silenciamiento de ambos genes en la mutación completa. Las flechas ubicadas en esta isla así como en cada gen simbolizan la orientación de la transcripción. Las líneas verticales en FMR1 representan los exones de este gen.")
Ubicación genómica y rol de FMR1 y FMR1-AS1. La barra gris representa la posición genómica (en kilobases [K]) de ambos genes en el cromosoma X desde el extremo p-terminal. La caja negra simboliza la isla CpG involucrada en el silenciamiento de ambos genes en la mutación completa. Las flechas ubicadas en esta isla así como en cada gen simbolizan la orientación de la transcripción. Las líneas verticales en FMR1 representan los exones de este gen.
La proteína FMRP tiene un máximo de 631 aminoácidos y contiene 2 motivos de unión a RNA, señales de localización nuclear y de exportación nuclear, y 2 zonas de interacción proteína-proteína46. Los niveles de expresión de FMRP varían según el tejido y el tipo de célula de un mismo tejido. Por ejemplo, en el ser humano se expresa ampliamente en epitelios y en el sistema nervioso central. Dentro de este último, se expresa en troncoencéfalo, estructuras derivadas del prosencéfalo y cerebelo, aunque en mayor medida en neuronas que en la glía. Finalmente, en las neuronas la proteína se concentra en el pericarion, dendritas proximales y en las sinapsis47.
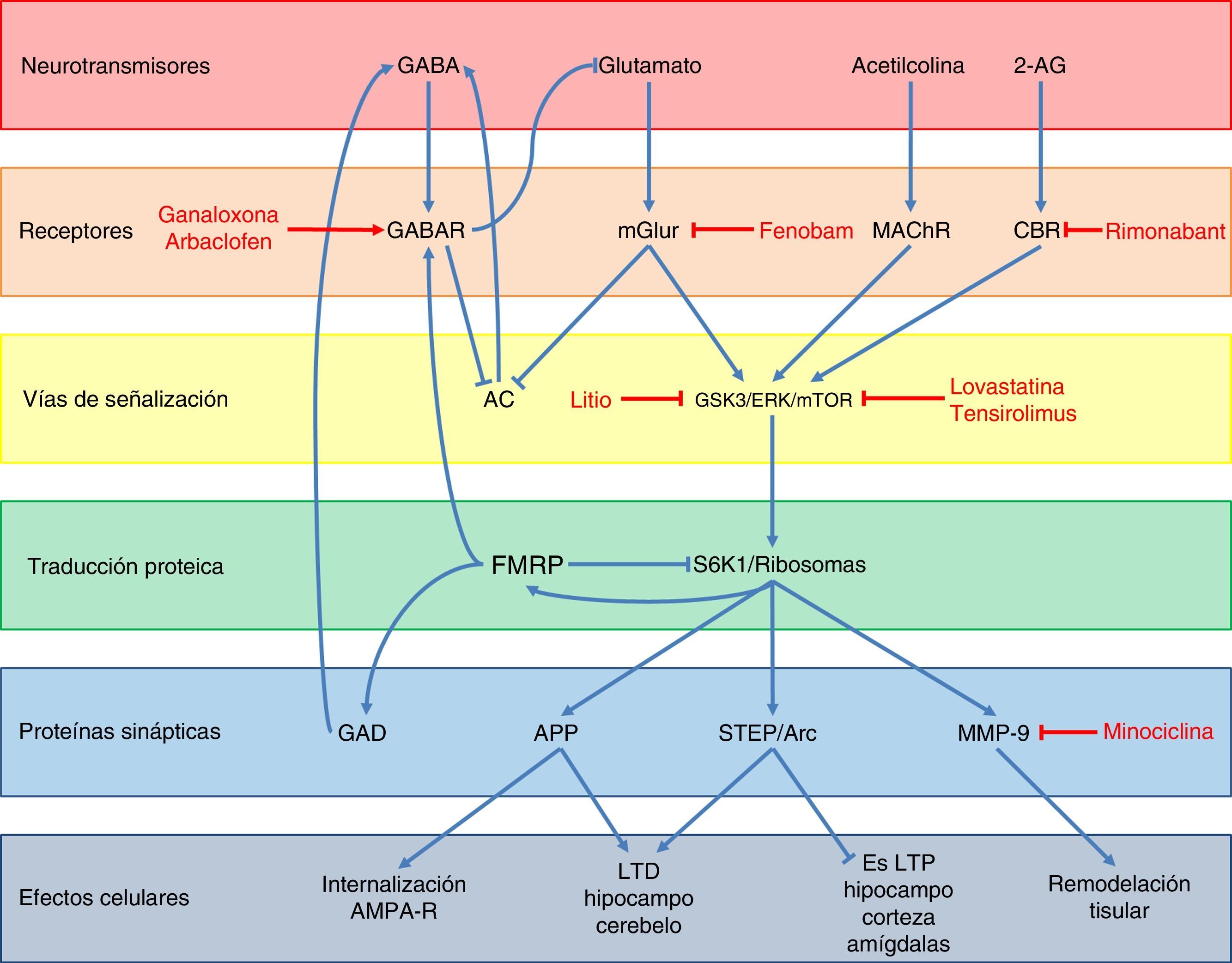
Con respecto a la función de la FMRP, se han descrito diversas vías por la cuales regularía la expresión de otros genes y la sinapsis neuronal (fig. 3)48. En condiciones normales, la FMRP es una proteína que se une a diversos mRNA49, interactúa con el ribosoma 80S50 y disminuye la actividad de la cinasa ribosomal S6K149, impidiendo de esta forma la traducción de diversas proteínas sinápticas como son la proteína precursora de amiloide (APP), la proteína tirosin-fosfatasa enriquecida en el estriado (STEP), la proteína Arc y la metaloproteinasa 9 (MMP-9)48,51,52. Dichas proteínas favorecen la internalización de receptores AMPA, la depresión a largo plazo (LTD) en el hipocampo y cerebelo, y a la remodelación tisular a nivel sináptico, mientras que a su vez disminuyen la potenciación a largo plazo (LTP) en el hipocampo, la corteza y las amígdalas48,49,51. Además, la FMRP favorece la expresión de receptores GABA tipo A (GABAR-A) y de la descarboxilasa de ácido glutámico (GAD), la enzima limitante en la síntesis de GABA48,53. Finalmente, todo esto se traduce en que la FMRP participa en la maduración de las espinas dendríticas y en la sinaptogénesis54, en el transporte y regulación del transporte de proteínas en la dendrita55, y actúa como un represor traduccional en la sinapsis, regulando los niveles de mRNA de proteínas involucradas en la estructura y función sináptica56, jugando un rol clave en la formación de patrones regulados de mRNA a nivel subcelular y temporal durante el desarrollo57.

Esquema de la neurobiología de la FMRP y los distintos blancos terapéuticos y fármacos usados en el SXF, según los componentes celulares con los cuales interactúa FMRP. A nivel de receptores, ganaloxona y arbaclofeno son agonistas de los receptores GABA-A y GABA-B, respectivamente; fenobam es un antagonista de los receptores mGluR5, mientras que rimonabant es un antagonista de los receptores endocanabinoides CB1R. Por otra parte, las dianas dentro de las vías de transducción de señales son GSK3, ERK y mTOR, los cuales son inhibidos por el litio, lovastatina y tensirolimus, respectivamente. Finalmente, minociclina inhibe la metaloproteinasa de matriz MMP-9.
La ausencia de FMRP lleva a diversas alteraciones en distintos sistemas de neurotransmisores. La principal y más estudiada es la desregulación de la vía del glutamato, particularmente a través de los receptores metabotrópicos (mGluR) y las proteínas río abajo de estos. Debido a la pérdida de la función reguladora de FMRP, se produce una sobreexpresión de APP, STEP, Arc y MMP-9, que son los efectores finales de la vía de mGluR (fig. 3)48,51. La sobreactivación de la vía glutamatérgica también se debe a la falta de contrarregulación por la vía de GABA debido a la subexpresión de receptores GABAR-A y la disminución en la síntesis de este neurotransmisor53. Este sistema además inhibe la liberación de glutamato, por lo que la falta de FMRP lleva indirectamente a una mayor activación de los mGluR48. Por otro lado, estos receptores glutamatérgicos también conducen a la inhibición de la adenilato ciclasa (AC)58 y una subsecuente disminución en la liberación de GABA. Por último, las vías endocanabinoide y de acetilcolina también participan en la mayor activación de la vía glutamatérgica al interactuar con las proteínas mTOR y ERK, las cuales son proteínas de transducción de señales río abajo de los mGluR, y que se ven activadas por los receptores canabinoides (CBR) y muscarínicos de acetilcolina (mAChR)48,59 (fig. 3). Todo esto lleva a una debilidad de las conexiones sinápticas60–63 y una mayor susceptibilidad a las convulsiones64.
En el rango de PM, el gen FMR1 se transcribe de manera eficiente y la expresión de FMRP es casi normal, pero a expensas de una pobre traducción del mRNA que se ve compensada por mayores niveles (entre 5-8 veces) de mRNA14,34. De esta forma, se produce una acumulación de inclusiones ubiquitina positiva en los núcleos de las neuronas y glías del SNC65 que contienen mRNA de FMR1 expandido y proteínas con motivos de unión a RNA que son secuestradas, afectando su función y produciendo neurodegeneración66. El número y tamaño de las inclusiones aumentan con la edad en paralelo con la progresión de la enfermedad en humanos67. Además, hay disfunción mitocondrial en fibroblastos y el tejido cerebral de portadores de PM, con incremento del estrés oxidativo y disminución de la traducción de proteínas mitocondriales68.
Finalmente, aún no está muy claro el rol de FMR1-AS1 en la patogénesis del SXF. Khalil et al.69 demostraron que el silenciamiento parcial de este lncRNA producía alteraciones en el ciclo celular y apoptosis, sin influir en la expresión de FMR1 y viceversa, sugiriendo un mecanismo independiente de este último gen. De esta forma, quedan aún muchos interrogantes en relación con la función de FMR1 y FMR1-AS1 (fig. 2). A pesar de esto, parece relevante resumir que el SXF se debería a una falta de expresión de ambos genes, mientras que los trastornos asociados a la PM se deberían a una sobreexpresión de mRNA tanto de FMR1 como de FMR1-AS1, lo que llevaría a degeneración celular.
Confirmación del diagnósticoLos exámenes para el estudio de mutaciones del gen FMR1 son sensibles y específicos, tanto para el diagnóstico de pacientes clínicamente afectados, como para portadores asintomáticos. Estos se realizan a partir de una muestra de sangre de la cual se extrae el ADN, y en un análisis directo del gen se determina el número de repeticiones CGG y el estado de metilación del locus70,71.
Actualmente existen diversos métodos moleculares para el análisis de las mutaciones en el locus FRAXA: 1. Los métodos basados en la reacción en cadena de la polimerasa (PCR) son en general técnicas rápidas, simples y de bajo costo especialmente si se aplican solo en hombres, pero que también permiten determinar el tamaño exacto de los alelos que se encuentran en los rangos de normalidad, zona gris, PM y alelos con MC incluso en mujeres72,73 los que, sin embargo, deben ser confirmados por Southern Blot70. 2. El estudio por Southern Blot permite el análisis directo del locus FRAXA y del gen FMR1, determinando además del tamaño de la expansión, el estado de metilación del gen y permite inferir a partir de este dato los niveles de expresión de la proteína. Estas técnicas están disponibles en Chile, pero actualmente sus costos no son cubiertos por las instituciones aseguradoras de salud71,74 (fig. 4). 3. Recientemente se han implementado metodologías basadas en PCR cuantitativo de tiempo real y denaturación sensible a metilación (MS-QMA) que permiten determinar el estado de metilación de otros sitios CpG dentro del gen FMR1 y que se correlacionan con el nivel de expresión de FMRP y el nivel cognitivo de los pacientes estudiados75.
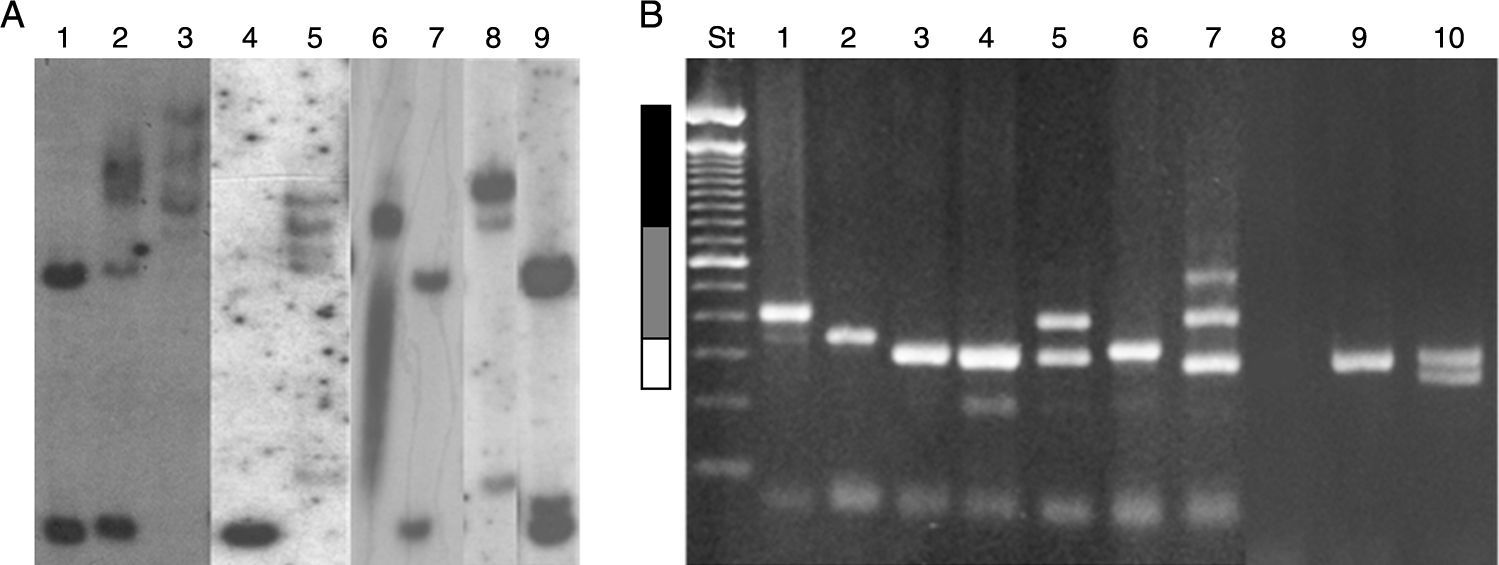
 Resultados obtenidos por Southern Blot. Carriles 1 y 7: mujer normal; carril 2: mujer MC; carril 9: mujer PM; carril 4: hombre normal; carril 3: hombre MC; carriles 5 y 8: hombres mosaico PM/MC; carril 6: hombre MC parcialmente desmetilado. B) Resultados obtenidos en el diagnóstico por PCR. Carriles 1 y 2: hombres con PM; carriles 3, 6 y 9 hombres normales; carril 4: mujer normal (homocigoto, confirmado por SB); carril 5: mujer PM; carril 7: mujer mosaico PM/MC; carril 8: hombre con MC; carril 10: mujer normal (heterocigota). St: estándar de peso molecular 100pb (invitrogen). Barra blanca: rango normal menor a 55 repeticiones CGG; barra gris: rango de PM entre 55-200 repeticiones CGG, y barra negra: MC mayor a 200 repeticiones CGG.")
Resultados moleculares en el diagnóstico de desórdenes de FMR1. A) Resultados obtenidos por Southern Blot. Carriles 1 y 7: mujer normal; carril 2: mujer MC; carril 9: mujer PM; carril 4: hombre normal; carril 3: hombre MC; carriles 5 y 8: hombres mosaico PM/MC; carril 6: hombre MC parcialmente desmetilado. B) Resultados obtenidos en el diagnóstico por PCR. Carriles 1 y 2: hombres con PM; carriles 3, 6 y 9 hombres normales; carril 4: mujer normal (homocigoto, confirmado por SB); carril 5: mujer PM; carril 7: mujer mosaico PM/MC; carril 8: hombre con MC; carril 10: mujer normal (heterocigota). St: estándar de peso molecular 100pb (invitrogen). Barra blanca: rango normal menor a 55 repeticiones CGG; barra gris: rango de PM entre 55-200 repeticiones CGG, y barra negra: MC mayor a 200 repeticiones CGG.
Se ha demostrado en modelos de ratones SXF que la exposición a un ambiente enriquecido puede mejorar la conducta, incluyendo materiales de nidificación, ejercicio físico y juguetes de diferentes texturas y colores76. Los pacientes con SXF parecen beneficiarse con terapia fonoaudiológica y ocupacional77. En general, es posible conseguir importantes logros en el manejo del comportamiento, trastornos conductuales y síntomas autistas de los niños X frágil, a partir de la implementación de técnicas específicas tanto en las instituciones educativas como en el ambiente familiar31.
Manejo farmacológicoEl tratamiento de los pacientes con SXF se puede categorizar en aquellos que van dirigidos a controlar los síntomas neuropsiquiátricos presentes y en aquellos que actúan sobre la base fisiopatológica de esta enfermedad.
Tratamiento de las manifestaciones clínicasEl manejo es básicamente de los síntomas y los trastornos prevalentes en SXF como son el TDAH, la ansiedad, los trastornos de conducta y las convulsiones. Los medicamentos más ampliamente usados son los psicoestimulantes para el manejo de la atención y la hiperactividad, los inhibidores de la recaptación de la serotonina para reducir la agresividad asociada a la ansiedad y los antipsicóticos atípicos para el manejo de la irritabilidad78,79. En niños menores de 5 años, se prefieren los agonistas alfa adrenérgicos como la clonidina y la guanfacina para el manejo del TDAH79. El uso de L-acetilcarnitina ha mostrado disminuir los síntomas de esta patología en estudios piloto controlados80. Ensayos realizados con sertralina en grupos pequeños de pacientes mostraron reducción de la ansiedad81 y un estudio realizado en 15 pacientes con aripiprazol mostró mejoría significativa de la irritabilidad78. Sin embargo, faltan ensayos clínicos aleatorizados doble ciego y con un mayor número de pacientes para validar definitivamente estas terapias.
Con respecto a las convulsiones, en la actualidad se prefiere el uso de carbamazepina o ácido valproico por sus escasos efectos adversos en estos pacientes y el buen control de las crisis epilépticas17,82. Tanto lamotrigina como levetiracetam han mostrado efectos aditivos potentes en aquellos pacientes de difícil control, presentando además la ventaja de producir mínimos efectos cognitivos79. Finalmente, se recomienda evitar el uso de fenitoína, fenobarbital o gabapentina, ya sea por sus efectos adversos o por la exacerbación de los problemas de conducta79.
Tratamientos experimentales basados en la fisiopatología del síndrome x frágil (SXF)Con los avances en el entendimiento de la neurobiología del SXF se han ido aplicando y desarrollando diversos fármacos que van a actuar en las vías de neurotransmisores involucradas en la patología. Al igual que en los mecanismos fisiopatológicos descritos en la figura 3, podemos agrupar estas terapias farmacológicas experimentales en aquellas que actúan a nivel de los receptores de neurotransmisores involucrados, en las proteínas de señalización intracelular río abajo de estos o en las proteínas sinápticas efectoras.
Dentro del primer grupo se encuentran los agonistas de los receptores GABA y distintos antagonistas de la vía glutamatérgica, con el fin de compensar su sobreactivación producida por el déficit de FMRP48 (fig. 3). Es así como el tratamiento con anticonvulsivantes agonistas de receptores GABA-A, como ganaxolona, pueden controlar las convulsiones y la ansiedad en SXF83, aunque también pueden aumentar la somnolencia84,85. El baclofen, agonista de receptores GABA-B, demostró ser eficaz en el tratamiento de la hiperactividad y las crisis convulsivas en ratones knockout (KO) de FMR186. En humanos, arbaclofen, un isómero de baclofen más potente en regular receptores GABA-B, disminuyó las conductas irritativas y mejoró la interacción social87,88, pero faltan estudios para evaluar su toxicidad y efecto a largo plazo. Por otro lado, estudios realizados con fenobam, un antagonista de receptor de glutamato (mGluR5), mostraron normalización en el fenotipo conductual y en las anomalías dendríticas del hipocampo en ratones KO de FMR148. Un ensayo preliminar, realizado en 12 pacientes adultos con SXF tratados con una dosis única de este fármaco, evidenció mejorías en la interacción social y la hiperactividad, pero presentaron leve sedación y aumento de las conductas ansiosas89. Actualmente se encuentran en curso ensayos clínicos con otros antagonistas de mGluR590. Con respecto a la vía endocanabinoide, se está probando en animales rimonabant, un antagonista del receptor canabinoide CB1 que impide la interacción con su ligando natural 2-araquidonoilglicerol (2-AG)48. El tratamiento agudo con este fármaco en ratones KO de FMR1 corrigió total o parcialmente la memoria de reconocimiento de objetos y la susceptibilidad a convulsiones audiogénicas, mientras que el tratamiento crónico mejoró la densidad de espinas dendríticas y disminuyó la señalización por mTOR en el hipocampo59.
Considerando las proteínas involucradas en la señalización intracelular río abajo de los receptores mGluR, se están analizando diversos fármacos que las inhiben48 (fig. 3). El litio, un inhibidor de la cinasa tipo 3 de la glicógeno sintasa (GSK3) y que está sobreactivada en ratones KO de FMR148, en general es bien tolerado y en un ensayo piloto con 15 pacientes demostró mejorar significativamente el comportamiento, las conductas adaptativas y la memoria verbal en jóvenes con SXF91. Por otra parte, lovastatina y tensirolimus, inhibidores de ERK y mTOR respectivamente, disminuyeron la síntesis de las proteínas involucradas en la LTD, los déficit de memoria y la susceptibilidad a convulsiones audiogénicas en ratones KO de FMR148.
Por último y con respecto a las proteínas sinápticas efectoras, también se están llevando a cabo estudios con inhibidores48. En ratones, la actividad inhibitoria de MMP-9 de minociclina mejoró significativamente su desempeño intelectual y disminuyó las conductas ansiosas a través de la maduración de las espinas dendríticas en el hipocampo92. En un estudio inicial en niños tratados con minociclina, la droga fue bien tolerada y habría disminuido la irritabilidad, los movimientos estereotipados, la hiperactividad y el lenguaje inapropiado93, pero está pendiente la realización de ensayos controlados.
Hasta la fecha no hay revisiones sistemáticas de los distintos tratamientos farmacológicos planteados debido al bajo número de pacientes, la corta duración de los tratamientos y la dificultad de sistematizar las escalas de evaluación. Las estrategias terapéuticas actuales han demostrado impacto en los síntomas, pero no mejoran el perfil cognitivo de los pacientes. En general, son los tratamientos combinados, farmacológicos y no farmacológicos los que han reportado mayores beneficios94.
Tratamiento de insuficiencia ovárica primaria relacionada al gen 1 del retardo mental del x frágil (FXPOI) y síndrome de temblor/ataxia asociado al X frágil (FXTAS)Manejo de la insuficiencia ovárica primaria relacionada al gen 1 del retardo mental del X frágil (FXPOI)Las mujeres con PM y FXPOI pueden presentar síntomas asociados a la menopausia, además del efecto sicológico de la pérdida prematura de su capacidad reproductiva. Aun cuando no existe un tratamiento específico, es importante considerar los beneficios de la psicoterapia95. Por otro lado, es importante destacar que la presencia de FXPOI no elimina la posibilidad de embarazos subsecuentes, por lo que las mujeres afectadas deberían ser derivadas a ginecología para evaluar sus posibilidades reproductivas. Además, debido a los bajos niveles séricos de estradiol y las consecuencias multisistémicas de esto, también deben ser evaluadas por endocrinología para considerar el uso de terapia de reemplazo hormonal39.
Manejo del síndrome de temblor/ataxia asociado al x frágil (FXTAS)El tratamiento farmacológico de los trastornos psiquiátricos asociados a PM es inespecífico y se basa en las intervenciones psicofarmacológicas convencionales. Para el tratamiento de los trastornos del ánimo, los medicamentos más utilizados son los inhibidores selectivos de la recaptación de serotonina95.
No hay un manejo específico para los trastornos del movimiento en FXTAS, pero algunos medicamentos pueden contribuir a disminuir la sintomatología. Para el temblor de intención, el propranolol y la primidona son los fármacos más utilizados. Algunos estudios demostraron mejoría con toxina botulínica, levetiracetam, clonazepam, clozapina, nadolol y nimodipino43. Para el tratamiento de la ataxia, además de la terapia física, algunos pacientes han mostrado mejoría con amantadina o buspirona96. El uso de antipsicóticos debe efectuarse con precaución, debido a la posibilidad de aumentar los trastornos del movimiento. La quetiapina se asocia a menor riesgo de efectos secundarios extrapiramidales43,95.
Aunque no existe una recomendación formal por alguna asociación, las personas adultas portadoras de PM deberían controlarse al menos anualmente los niveles séricos de la hormona estimulante del tiroides, tiroxina libre y triyodotironina, de manera de pesquisar y tratar precozmente el hipotiroidismo97.
Hasta el momento existe escasa evidencia para recomendar algún fármaco para el manejo de la demencia en FXTAS. Sin embargo, recientemente se mostró que el tratamiento con memantina durante un año mejoró significativamente la memoria verbal en pacientes con FXTAS98, lo cual entrega bases para nuevos ensayos clínicos con este medicamento.
Por último, hasta la fecha no se han descrito estudios experimentales con RNA de interferencia que contrarresten los niveles elevados de mRNA de FMR1 presentes en pacientes con la PM. Sin embargo, es un blanco terapéutico a estudiar tomando en cuenta la experiencia con este tipo de oligonucleótidos en la distrofia miotónica tipo 137.
Asesoramiento genéticoCuando se sospecha que un individuo puede padecer de SXF, es muy relevante explicar a la familia las implicaciones que el estudio molecular puede tener, no solo para el paciente, sino para el resto de los miembros, y la importancia de derivarlo a un genetista clínico42. El genetista recopilará los antecedentes de la familia y facilitará el acceso a estudio de los familiares, informándoles a tiempo del riesgo que conlleva ser portador de una mutación y contribuir en el proceso de planificación familiar42,99.
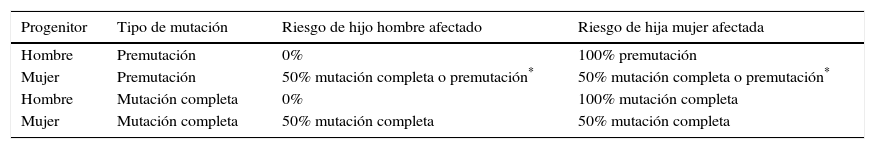
El riesgo de transmitir la mutación a la descendencia depende del género y del número de repeticiones. Los hombres premutados trasmiten la PM a todas sus hijas y a ninguno de sus hijos. Las mujeres portadoras de PM tienen un riesgo de un 50% de transmitir el alelo alterado en el rango de PM o MC a cualquiera de sus hijos. Las mujeres con MC tienen un riesgo de un 50% de que sus hijos(as) hereden la MC. En el caso de los hombres con MC, sus hijos no heredan la mutación y sus hijas pueden heredar la MC o una PM por contracción de la expansión99 (tabla 3).
Riesgo de transmitir la mutación a la descendencia
| Progenitor | Tipo de mutación | Riesgo de hijo hombre afectado | Riesgo de hija mujer afectada |
|---|---|---|---|
| Hombre | Premutación | 0% | 100% premutación |
| Mujer | Premutación | 50% mutación completa o premutación* | 50% mutación completa o premutación* |
| Hombre | Mutación completa | 0% | 100% mutación completa |
| Mujer | Mutación completa | 50% mutación completa | 50% mutación completa |
Todos los familiares en riesgo de portar una PM o MC deben ser evaluados para detectar problemas neurológicos, emocionales y endocrinos. Las familias pueden ser invitadas a participar en asociaciones de apoyo, especialmente de las agrupaciones de padres que se han ido creando en cada país. Por otro lado, es necesario contar con datos de la población hispánica y latinoamericana con respecto al número de interrupciones AGG en mujeres portadoras de PM y cómo estas han incidido en la probabilidad de expansión en la descendencia, ya que esta es una recomendación de las nuevas guías clínicas españolas de diagnóstico y manejo de los TASXF con base en la evidencia que se ha obtenido de población estadounidense40,100.
Existen estudios de pesquisa poblacional de SXF, incluyendo diagnóstico prenatal, detección en poblaciones con compromiso neurológico y tamizaje neonatal101. Aunque aún existen controversias, el diagnóstico precoz a través de la pesquisa neonatal permitiría una intervención temprana, el asesoramiento a la familia y la toma de decisiones informadas de los padres102. Esto disminuiría posiblemente la aparición de nuevos casos, en la medida que el diagnóstico se realice antes del nacimiento de un segundo hijo afectado (fig. 1).
ConclusionesEl SXF se asocia a un amplio espectro de manifestaciones clínicas, que van desde el fenotipo clásico de la MC a las manifestaciones neurológicas y síntomas neuropsiquiátricos de la PM37. Esta gama de presentaciones y su alta frecuencia en la población hacen que la mayoría de los médicos tengan la posibilidad de encontrarse en su práctica clínica con pacientes afectados por SXF. El estudio molecular del gen FMR1 debe ser considerado dentro del estudio de pacientes con retraso del desarrollo sicomotor, DI, autismo, mujeres con menopausia precoz, adultos con marcha atáxica o temblor de intención, parkinsonismos, neuropatía periférica, demencia y trastorno ansioso o depresión, y especialmente si hay antecedentes familiares de DI y/o autismo99,103.
Los síntomas psiquiátricos de los pacientes premutados pueden ser una manifestación primaria del estado de PM, por lo que no se deben atribuir exclusivamente al estrés de la crianza de un niño discapacitado, aun cuando dicho estrés siempre está presente39.
Los exámenes moleculares disponibles y el asesoramiento genético de la familia, incluyendo la confección de una genealogía ampliada, son esenciales para establecer quiénes están en riesgo de tener mutaciones en FMR1, de manera de determinar su pronóstico y riesgo individual de transmitir la enfermedad a la descendencia99.
Los avances en la identificación de las bases moleculares del SXF pueden servir como modelo para comprender las causas de las enfermedades neuropsiquiátricas, para aclarar los mecanismos involucrados en los síntomas neurológicos y psiquiátricos que presentan otras enfermedades genéticas, y probablemente conducirán al desarrollo de tratamientos cada vez más específicos48.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

