




Las degeneraciones lobares frontotemporales (DLFT) son un grupo de patologías moleculares que se definen en función de la proteína acumulada en el sistema nervioso central. La demencia frontotemporal variante conductual (DFT vc) es el síndrome clínico de presentación más frecuente. Los avances realizados en los últimos años han contribuido a un mayor conocimiento de esta entidad, que puede ser el modo de presentación de diferentes enfermedades neurodegenerativas.
DesarrolloSe revisa la correlación entre clínica, patología y genética de las DLFT, en especial de la DFT vc, así como los principales biomarcadores de la enfermedad. La anatomía patológica de la DFT vc es muy variada, sin mostrar asociación significativa con ningún subtipo histopatológico concreto. Entre los biomarcadores disponibles, destacan la neuroimagen anatómica y funcional, los biomarcadores analíticos y la genética. Se están diseñando fármacos dirigidos contra dianas moleculares concretas implicadas en la patogenia de las DLFT.
ConclusionesLa DFT vc es una causa frecuente de demencia. De entre todas las variantes clínicas de las DLFT, es en la que resulta más difícil establecer una relación clínico-patológica. El uso de biomarcadores puede ayudar a predecir la anatomía patológica subyacente, lo que junto al desarrollo de fármacos ligando-específicos ofrece nuevas posibilidades terapéuticas.
Lobar frontotemporal degeneration (FTLD) encompasses a group of molecular disease defined by the deposition of an abnormal protein in the central nervous system. Behavioural variant frontotemporal dementia (bvFTD) is the most frequent clinical presentation of FTLD. The past two decades of research have contributed to a better understanding of this entity, which may be the first manifestation in many different neurodegenerative disorders.
DevelopmentWe reviewed correlations between clinical, pathological, and genetic findings and the main disease biomarkers of FTLD, with particular interest in bvFTD. Anatomical pathology findings in FTLD are heterogeneous and the syndrome is not associated with any one specific histopathological type. Promising available biomarkers include structural and functional neuroimaging techniques and biochemical and genetic biomarkers. Disease-modifying drugs designed for specific molecular targets that are implicated in FTLD pathogenesis are being developed.
ConclusionsBvFTD is a frequent cause of dementia. Of all the clinical variants of FTLD, behavioural variant is the one in which establishing a correlation between clinical and pathological signs is the most problematic. A biomarker evaluation may help predict the underlying pathology; this approach, in conjunction with the development of disease-modifying drugs, offers new therapeutic possibilities.
El término degeneración lobar frontotemporal (DLFT) es un concepto patológico que engloba a un grupo de enfermedades o, lo que es lo mismo, de patologías moleculares que se clasifican en función de la proteína que se deposita en el sistema nervioso central. En 2007, Cairns et al.1 establecieron 4 subtipos principales en función de la proteína acumulada: taupatías (DLFT-tau), ubiquitinopatías (DLFT-U), demencia sin cambios histopatológicos específicos y enfermedad por inclusión neuronal de filamentos intermedios. Posteriormente, tras los nuevos avances en patología y biología molecular, esta clasificación ha experimentado algunas modificaciones. Actualmente, se prefiere clasificar a las DLFT en 3 grupos patológicos principales: DLFT asociada a tau (DLFT-tau), DLFT asociada a la proteína fijadora de ADN TAR-43 (DLFT-TDP) y DLFT asociada a la proteína de fusión en sarcoma (DLFT-FUS). Solo un porcentaje extremadamente pequeño de casos escaparía de esta clasificación (DLFT-otros)2. Cada uno de los principales grupos patológicos se subdivide en varias entidades o variantes patológicas en función de la distribución y la morfología de las inclusiones proteicas y las características histológicas específicas (tabla 1). Estos grupos originan 6 entidades sindrómicas bien definidas: las 3 variantes clínicas de la demencia frontotemporal (demencia frontotemporal variante conductual [DFT vc], afasia primaria progresiva no fluente [APPNF] y demencia semántica [DS]), la demencia frontotemporal asociada a enfermedad de la motoneurona (DFT-EMN), el síndrome de parálisis supranuclear progresiva (SPSP) y el síndrome corticobasal (SCB).
Clasificación histopatológica y biomolecular de las degeneraciones lobares frontotemporales
| Tipo y subtipo histopatológico | Proteína | Características histopatológicas |
|---|---|---|
| DLFT-tAU | ||
| Enfermedad de Pick | tau 3R | Inclusiones citoplasmáticas redondeadas argirófiras (cuerpos de Pick) Células no piramidales giro dentado y área CA1 del hipocampo Neuronas piramidales lóbulos frontales y temporales (capas ii y vi) |
| Degeneración corticobasal | tau 4R | Ovillos redondeados en neuronas y glía con predominio cortical Depósito de tau en astrocitos en forma de corona («placa glial») Depósitos en oligodendrocitos («coiled bodies») |
| Parálisis supranuclear progresiva | tau 4R | Ovillos redondeados en neuronas y glía con predominio subcortical Astrocitos «en penacho» |
| Demencia con granos argirófiros | tau 4R | Depósito en espinas dendríticas Depósitos en oligodendrocitos («coiled bodies») |
| Taupatía multisistémica | tau 4R | Neocórtex (lóbulos frontales, temporales y parietales) Ganglios basales y tronco del encéfalo |
| Demencia con predominio de ovillos | tau 3R/4R | Ovillos neurofibrilares corticales Ausencia de placas de amiloide |
| DLFT ligada mutación del gen MAPT | tau 3R, 4R o 3R/4R | Heterogeneidad marcada |
| DLFT-U | ||
| DLFT-TDP | ||
| Tipo 1 | TDP-43 | Abundantes NCI y DN NII variables («cat eyes») |
| Tipo 2 | TDP-43 | Abundantes DN Pocos o ningún NCI o NII |
| Tipo 3 | TDP-43 | Numerosos NCI Escasos NII/DN |
| Tipo 4 | TDP-43 | Numerosos NII NCI y DN infrecuentes |
| DLFT-FUS | ||
| Enfermedad neuronal por inclusión de filamentos intermedios | FUS | NCI positivos para FUS, filamentos intermedios y ubiquitina Visibles en H&E |
| Enfermedad por inclusión de cuerpos basófilos | FUS | NCI positivos para FUS y ubiquitina, negativos para TDP-43, tau y filamentos intermedios Basófilos en H&E |
| DLFT atípica con inclusiones ubiquitina positivas | FUS | NCI positivos para FUS y ubiquitina, negativos para TDP-43, tau y filamentos intermedios |
| Otras-DLFT | ||
| DLFT sin inclusiones | ¿? | Pérdida neuronal y gliosis frontotemporal No evidencia de inclusiones |
| DLFT con imnunorreactividad para proteínas del sistema ubiquitina proteasoma | Ubi | Inclusiones positivas para ubiquitina o p62 Negativas para tau, α-sinucleína, α-internexina, TDP-43 y FUS |
| No DLFT | ||
| EA | tau 3R y 4R β-amiloide | Placas neuríticas positivas para β-amiloide Ovillos neurofibrilares positivos para tau 3R y 4R |
Los subtipos de DLFT-TDP 1-4 se han clasificado según el esquema de Mackenzie.
EA: enfermedad de Alzheimer; DLFT-FUS: degeneración lobar frontotemporal asociada a la proteína de fusión en sarcoma; DLFT-tau: degeneración lobar frontotemporal asociada a depósito de la proteína tau; DLFT-TDP: degeneración lobar frontotemporal asociada a depósito de la proteína fijadora de ADN TAR 43; DLFT-U: degeneración lobar frontotemporal asociada a depósito de ubiquitina; NCI: inclusiones citoplasmáticas neuronales; NII: inclusiones intranucleares neuronales; Ubi: ubiquitina.
Existe un pequeño porcentaje de casos con clínica de DFT que presentan patología de enfermedad de Alzheimer (EA)3. Este hallazgo es más frecuente en la afasia primaria progresiva (APP) logopénica4, aunque también se ha descrito en algunos casos de DS (10%)5 y DFT vc (5-7%)5-7. Los casos de EA con diagnóstico ante mortem de DFT vc se han denominado «EA variante frontal»8.
En el presente estudio, pretendemos revisar la correlación clínico-patológica y biomolecular de las DFT, en particular de la DFT vc, así como la utilidad de los biomarcadores para aproximarse a la «enfermedad» a partir de la clínica.
Correlación clínico-patológica y biomolecular de las degeneraciones lobares frontotemporalesClásicamente, se ha considerado a las DLFT como un grupo heterogéneo de enfermedades neurodegenerativas en las que resultaba extraordinariamente difícil establecer una relación entre la presentación clínica y el proceso patológico subyacente. Tampoco se creía posible deducir la naturaleza de las manifestaciones clínicas conociendo el diagnóstico patológico. Los avances en neuropatología, bioquímica, biología molecular y genética han permitido establecer no solo una correlación clinicopatológica, sino también clínica, histológica, biomolecular y genética.
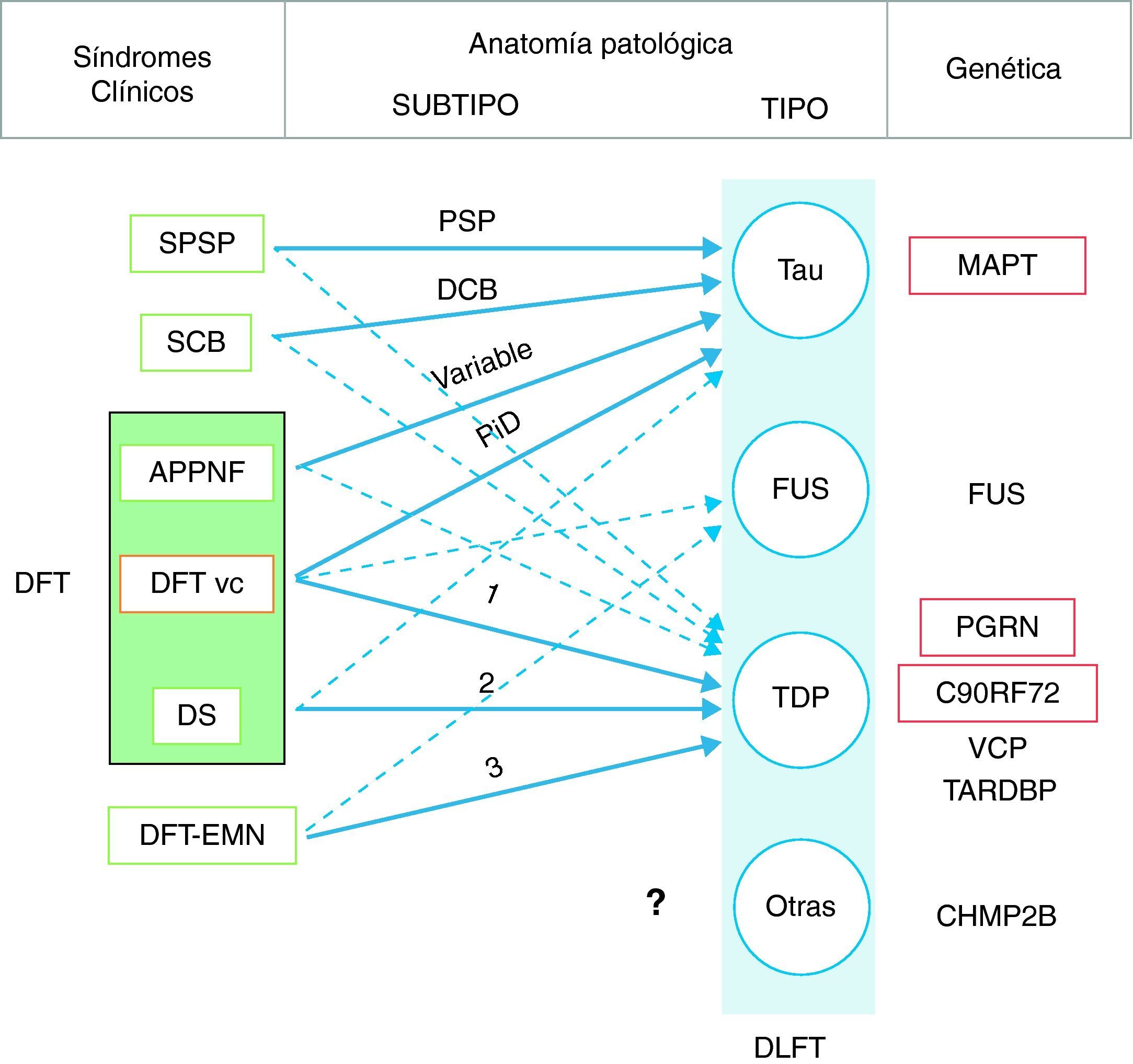
En 2011 Josephs et al.9 realizaron una revisión de varios estudios clínico-patológicos10-17 realizados en la última década en 6 centros diferentes de Estados Unidos, Canadá, Reino Unido y Australia, que reunían un total de 544 casos de DLFT. Observaron que la DFT vc mostraba patología DLFT-tau y DLFT-TDP en una proporción similar. Casi todos los casos de SPSP y SCB presentaban patología DLFT-tau, mientras que el 100% de los casos de DFT-EMN tenían patología DLFT-TDP. El 83% de los pacientes con DS asociaba patología DLFT-TDP, mientras que la APPNF estaba asociada fundamentalmente a DLFT-tau (70%). Al analizar por grupos (DLFT-TDP y DLFT-tau), encontraron una asociación significativa entre el fenotipo clínico y el subtipo histopatológico, excepto en el caso de la APPNF y el subtipo DLFT-tau, en el que la asociación no alcanzó la significación estadística. Los pacientes con DFT vc y patología tau, se asociaron a enfermedad de Pick (PiD) en el 70% de los casos y los que presentaron patología TDP mostraron asociación con el subtipo 1 de la clasificación de Mackenzie et al.18. No obstante, la asociación encontrada entre la DFT vc y el subtipo histopatológico fue débil (fig. 1).

Correlación clínica, molecular y genética de las degeneraciones lobares frontotemporales.
Las flechas indican la relación entre los síndromes clínicos y la patología subyacente, las de trazo continuo señalan las asociaciones más fuertes. Asimismo se indica el subtipo histopatológico más frecuente en cada caso. DFT: demencia frontotemporal; DLFT: degeneración lobar frontotemporal; SPSP: síndrome de parálisis supranuclear progresiva; SCB: síndrome corticobasal; APPNF: afasia primaria progresiva no fluente; DFT vc: demencia frontotemporal variante conductual; DS: demencia semántica; DFT-EMN: demencia frontotemporal asociada a enfermedad de la motoneurona; PSP: parálisis supranuclear progresiva; DCB: degeneración corticobasal; PiD: enfermedad de Pick; MAPT: microtubule associated protein tau; FUS: proteína de fusión en sarcoma; PGRN: progranulina; VCP: valosin containing protein; TARDBP: proteína fijadora de ADN TAR 43; CHMP2B: charged multivesicular body protein 2B. Los subtipos DLFT-TDP 1-3 se han clasificado según el esquema de Mackenzie.
En algunos casos de DFT vc, existen determinados rasgos clínicos que orientan a un subtipo histopatológico específico19. Existe un subtipo clínico que se caracteriza por cambios conductuales y de personalidad, en el que destacan la hipersexualidad, la hiperfagia, la estereotipia y los comportamientos obsesivos, que presenta una fuerte asociación a patología DLFT atípica con inmunorreactividad exclusiva a ubiquitina (aDLFT-U)20,21. Estos casos presentan además un comienzo muy temprano (edad media al inicio en torno a los 40 años) y se caracterizan por una marcada atrofia estriatal22,23. Por otra parte, la coexistencia de síntomas psicóticos o de signos de enfermedad de la motoneurona (EMN) orienta a patología DLFT-TDP tipo 324 y la apatía como rasgo dominante a DLFT-TDP tipo 1 asociada a mutación de la PGRN25,26. La coexistencia de desinhibición marcada y alteración del eje semántico del lenguaje sugiere patología tau asociada a mutación de«microtubule-associated protein tau» (MAPT)14,26,27. La presencia de parálisis supranuclear de la mirada vertical se asocia a patología tau, concretamente a degeneración corticobasal (DCB) o, en menor medida, a parálisis supranuclear progresiva (PSP)28.
Aunque la correlación clínico-patológica de la DLFT-FUS ha sido menos estudiada29-36, parece que también existe una asociación entre el fenotipo clínico y el subtipo de patología DLFT-FUS. La enfermedad por inclusión neuronal de filamentos intermedios se asoció a 3 síndromes, el más frecuente la DFT vc (menos frecuente DFT-EMN y SCB/esclerosis lateral primaria). La enfermedad con cuerpos de inclusión basófilos se asoció a DFT vc, DFT-EMN, EMN y esclerosis lateral amiotrófica (ELA) juvenil. La aDLFTU se asoció con fuerza a la DFT vc. No existen estudios clinicopatológicos acerca de la DLFT sin inclusiones y la DLFT con inclusiones citoplasmáticas con inmunorreactividad para proteínas del sistema ubiquitina proteasoma y negatividad para TDP-43 (DLFT-UPS).
Aunque durante los primeros 4 años suele predominar un síndrome en concreto, es habitual que con la evolución de la enfermedad los síndromes clínicos que integran las DFT se solapen37. Kertesz et al.38 estudiaron de forma prospectiva a 262 pacientes con criterios clínicos de DFT. Durante el seguimiento, constataron la aparición de un segundo e incluso un tercer síndrome clínico. Los pacientes con DFT vc desarrollaron hasta en un 50% de los casos una APPNF, en un 22,5% un SCB/SPSP, y en torno a un 16% una DS. Algo más del 50% de las APPNF desarrollaron una DFT vc y un tercio un SCB/SPSP. El resto de APPNF no evolucionó a ningún otro síndrome. Tres cuartas partes de los pacientes con DS desarrollaron una DFT vc y el resto no evolucionó a ningún otro cuadro clínico. Los casos con SCB/SPSP desarrollaron una DFT vc (50%) o una APPNF (50%).
Por tanto, dado que es posible que un mismo paciente manifieste diferentes expresiones clínicas a medida que la enfermedad progresa, podría afirmarse que un mismo tipo histopatológico es capaz de generar la aparición de distintos cuadros clínicos por mecanismos etiológicos o neuropatológicos desconocidos.
Correlación clínico-patológica de las variantes genéticas de las degeneraciones lobares frontotemporalesAunque la mayoría de los casos de DLFT son esporádicos, existe una pequeña proporción asociada a determinadas mutaciones genéticas. Actualmente, se conocen 7 genes implicados en casos de DLFT familiar: MAPT, progranulina (PGRN), C9ORF72, «valosin containing protein-1» (VCP-1), «charged multivesicular body protein 2B» (CHMP2B), proteína fijadora de ADN TAR-43 (TARDBP) y proteína de fusión en sarcoma (FUS). Las mutaciones en los genes MAPT, PGRN y C9ORF72 explican la mayoría de los casos de DFT con herencia autosómica dominante.
Las mutaciones de MAPT39 se asocian típicamente a patología DLFT-tau, sin existir un patrón histológico concreto40, pudiendo presentarse histológicamente como PiD, PSP y DCB41. Los pacientes con mutaciones del gen MAPT suelen presentar un fenotipo de DFT vc, en ocasiones con clínica extrapiramidal asociada27. Característicamente, presentan una marcada atrofia simétrica temporal anterior medial y orbitofrontal42,43, por lo que no es infrecuente que se manifiesten como una DS con rasgos de DFT vc (o DFT vc con alteración de los aspectos semánticos del lenguaje). La desinhibición es un rasgo especialmente frecuente en este grupo de pacientes23,26,27.
Los casos familiares asociados a mutación del gen de la PGRN presentan un patrón neuropatológico típico, con una marcada atrofia a nivel frontal, núcleo caudado, sustancia negra y región medial del tálamo. A diferencia de las mutaciones de MAPT, que se asocian a tau, se asocian a depósito de TDP-43, especialmente al tipo 1 de las DLFT-TDP según la clasificación de Mackenzie18, siendo las inclusiones intranucleares neuronales lenticulares o con forma de «ojo de gato» el hallazgo histológico más llamativo44-46.
Clínicamente, la expresión de las mutaciones en la PGRN es muy variable, no solo entre familias sino entre los componentes de la misma familia. La edad al inicio es también muy variable, incluso en la misma familia47. Pueden presentarse como un SCB48-51, APPNF23,51,52 y DFT vc48, en la que la apatía es un rasgo prominente25. La asociación de EMN es excepcional53. En los pacientes portadores de mutaciones de la PGRN, incluso en sujetos asintomáticos, los niveles plasmáticos de PGRN son muy bajos47,54-56.
La expansión del hexanucleótido GGGGCC en el primer intrón del gen C9ORF72 no solo es la mutación conocida más frecuentemente asociada a ELA y DFT-EMN, sino la segunda mutación más frecuente en la DLFT, después de las mutaciones de la PGRN. Según el estudio de DeJesus-Hernandez et al.57, se asocia al 12% de los casos familiares de DFT y al 22,5% de ELA, aunque se ha descrito que en poblaciones nórdicas esta prevalencia es algo mayor, encontrándose en el 46% de los casos de ELA familiar, el 21,1% de ELA esporádica y el 29,3% de DFT de la población finlandesa58.
Las mutaciones del gen C9ORF72, asociadas a patología DLFT-TDP, se pueden manifestar clínicamente como DFT vc, ELA o DFT-EMN. La variabilidad de presentación, incluso en una misma familia, es muy amplia. En el estudio de DeJesus-Hernandez, et al. 57, el 26,9% de los casos de DLFT presentaban ELA asociada y más del 30% tenía familiares con ELA.
Las mutaciones en el gen VCP-1 se asocian a una rara entidad autosómica dominante, que cursa con miopatía (90%), enfermedad de Paget ósea y menos frecuentemente DFT (30%), que suele aparecer años después del inicio de la clínica muscular59. De acuerdo con la clasificación histopatológica de las DLFT-TDP, se correspondería con el subtipo 4 de Sampathu y Mackenzie60. En la biopsia muscular de forma típica, aparecen los cuerpos de inclusión miopáticos.
Las mutaciones de CHMP2B, que hasta la fecha se han identificado únicamente en 5 familias, se asocian típicamente a DFT vc, con o sin clínica piramidal o extrapiramidal asociada61,62. En fases tardías, pueden aparecer mioclonías61. Únicamente en 2 casos se ha descrito EMN asociada63. Desde el punto de vista neuropatológico, se asocian a DLFT-UPS.
Las mutaciones en los genes TARDBP y FUS se asocian fundamentalmente a casos familiares de ELA, y son muy raras como causa de DFT64-67.
La relación de los diferentes fenotipos clínicos y la patología subyacente en los casos genéticos difiere respecto a la que se encuentra en los casos esporádicos. Por ejemplo, en los casos esporádicos la APPNF y el SCB se asocian preferentemente a DLFT-tau mientras que en los casos familiares se asocia con mayor frecuencia a DLFT-TDP (especialmente tipo 1). La DFT vc esporádica o familiar se asocia indistintamente a DLFT-tau o DLFT-TDP. La DS y el SPSP son casi siempre esporádicos.
Biomarcadores de neuroimagenNeuroimagen estructural: resonancia magnética cerebralLas enfermedades neurodegenerativas se asocian a atrofia cerebral progresiva, que se puede medir y cuantificar por medio de técnicas de neuroimagen estructural, como la RM cerebral. La cuantificación de la atrofia por medio de técnicas de neuroimagen ha mostrado ser un biomarcador útil en otras enfermedades neurodegenerativas68,69.
Existe una relación bien establecida entre los diferentes síndromes de DFT y el patrón de atrofia. En general, la DFT vc se asocia a atrofia que afecta predominantemente al lóbulo frontal, la ínsula, el cíngulo anterior y lóbulo temporal anterior, de forma asimétrica (predominio en el hemisferio no dominante). La DS se asocia con atrofia asimétrica que afecta a la región anteroinferior de los lóbulos temporales y la APPNF a atrofia asimétrica (más marcada en el hemisferio dominante) del córtex perisilviano anterior70-72.
Lindberg et al.73 analizaron los volúmenes de 9 regiones corticales de interés de 61 individuos (27 controles, 12 DFT vc, 9 APPNF y 13 DS) y compararon los hallazgos en las diversas entidades clínicas. Observaron que era posible discriminar entre unos síndromes y otros con sensibilidad y especificidad relativamente altas. La atrofia temporal frente a la frontal fue el patrón más útil para diferenciar la DS de los otros dos subtipos clínicos. La lateralidad de la atrofia, derecha frente a izquierda, fue el parámetro más útil para discriminar entre DFT vc y APPNF. A pesar de ello, hay que tener en cuenta que existe cierta heterogeneidad y solapamiento en los hallazgos neuroanatómicos y que en las fases precoces la RM cerebral puede no mostrar alteraciones.
Las alteraciones cerebrales que se encuentran en la DFT vc no solo se limitan al córtex cerebral. La atrofia también afecta a varias regiones cerebrales subcorticales, incluyendo la amígdala, el hipocampo, el caudado, el estriado, el putamen, el tálamo y el hipotálamo74,75. De hecho, la atrofia de la amígdala puede ser un marcador útil para diferenciar la DFT vc de la EA74. Asimismo, Chao et al.76 observaron que la reducción del volumen de la sustancia blanca frontal es un marcador de la reducción del volumen de la sustancia gris adyacente en pacientes con DFT vc y que diferentes tipos de DFT asociaban distintos patrones de atrofia de la sustancia blanca.
Cambios precoces, evolución y estadificación de la demencia frontotemporal variante conductualEl desarrollo de métodos automatizados cuantitativos, como la morfometría basada en vóxeles y el mapeo del grosor cortical, ha sido crucial para la detección de atrofia selectiva del cíngulo anterior y cortezas frontal e insular en estadios precoces de DFT vc71,77,78, que es diferente del patrón de atrofia que se observa en otras variantes de DFT y otras demencias, como la EA79. No obstante, en fases clínicas precoces puede no haber alteraciones macroscópicas, aunque se sabe que la patología molecular cerebral está presente años antes de la aparición de los síntomas (fase preclínica). La tasa anual de atrofia cerebral global en la DFT vc alcanza el 8%, casi el doble de la que se observa en la EA. Algunos pacientes con DFT vc pueden presentar tasas menores (0,3% por año), similares a los que se ven en individuos sanos80. Es posible que esta cifra tan baja se deba a la inclusión de fenocopias entre los casos.
La progresión de la atrofia cerebral en la DFT ha sido estudiada mediante el análisis del patrón de atrofia en pacientes con diferente duración de la enfermedad81. En fases iniciales la atrofia afecta generalmente a regiones mesioorbitarias del lóbulo frontal. Posteriormente, se afectan el polo temporal, la formación hipocampal, el córtex frontal dorsolateral y los ganglios basales. Este patrón de progresión se ha correlacionado con el volumen de regiones corticales y subcorticales y con la pérdida neuronal subyacente82,83. En consecuencia, el patrón de atrofia es útil para la estadificación de la gravedad de la enfermedad84,85. Basándose en estos hallazgos, Kipps et al.86 propusieron una escala visual de valoración mediante RM cerebral.
Seeley et al.77 llevaron a cabo un estudio de casos y controles en pacientes con DFT vc en diferentes estadios evolutivos. Clasificaron los casos en 3 grupos en función de la puntuación en la escala Clinical Dementia Rating (CDR 0,5, CDR 1 y CDR 2-3). En el grupo con afectación leve (CDR 0,5), se evidenció atrofia bilateral aunque asimétrica (predominio derecho), que afectaba a diversas regiones del lóbulo frontal (rostromedial, polo frontal, dorsolateral y orbitofrontal), cíngulo anterior, ínsula anterior, hipocampo y áreas subcorticales (estriado ventral y tálamo dorsomedial). A mayor puntuación CDR, la atrofia se hacía más extensa en dichas áreas, especialmente en el lóbulo frontal, y se extendía a áreas más posteriores, como la ínsula posterior y los lóbulos temporal y parietal. Otro estudio realizado sobre casos de DFT vc con confirmación patológica mostró similares resultados87
.
Estudios posteriores sugieren que estas áreas afectadas en estadios tempranos de la enfermedad (frontal-ínsula-cíngulo anterior) forman parte de un circuito neuronal estructural y funcionalmente diferenciado, denominado «Salience network» (SN). El sustrato anatómico de este circuito neural son las células de von Economo, una población neuronal única, que probablemente tiene un papel fundamental en la cognición social88,89. En este sentido, en un estudio con RM cerebral funcional se observó que la gravedad clínica de la DFT vc se correlacionaba con el grado de alteración del circuito SN derecho90. No obstante, dada la heterogeneidad patológica de la DFT vc, no está claro si este circuito neural está alterado por igual en todos los subtipos histopatológicos.
Correlación entre anatomía y subtipo patológicoMás interesante incluso, resulta el potencial de las técnicas de neuroimagen para aproximarse a la patología subyacente al síndrome.
Los primeros estudios realizados ofrecieron resultados contradictorios; unos mostraron diferencias entre pacientes con DLFT-tau y DLFT-U91, mientras que otros mostraron patrones de atrofia muy similares92. En 2009, Whitwell et al.93 analizaron mediante morfometría la atrofia cortical regional de una cohorte de 66 pacientes con diagnóstico clínico de DFT vc y observaron que la mitad de los pacientes presentaba una atrofia predominantemente frontal, mientras que en la otra mitad predominaba la atrofia temporal. Así pues, establecieron 4 subtipos anatómicos: frontal dominante, frontotemporal, temporofrontoparietal y temporal dominante. Los pacientes con patrón de atrofia frontal dominante y frontotemporal presentaron predominantemente afectación del lóbulo frontal (regiones orbitofrontal, mesial frontal y dorsolateral), en el primero bastante restringida y en el segundo también temporal, aunque en menor grado. Los pacientes con patrón temporofrontoparietal se caracterizaron por atrofia temporoparietal y medial frontal. El subtipo más infrecuente fue el temporal dominante, atrofia restringida a la región inferior y medial del lóbulo temporal, que en todos los casos se asoció a afectación predominantemente derecha y en el 83% a mutaciones del gen MAPT. Las mutaciones de la PGRN, a diferencia de las anteriores, no se asociaron en ningún caso al subtipo temporal dominante, lo que indica que el patrón anatómico podría ser útil para distinguir entre mutaciones de MAPT y PGRN. Los subtipos predominantemente temporales presentaron peores resultados en los test de lenguaje y memoria, y los de predominio frontal peores resultados en los de función ejecutiva, aunque no de forma estadísticamente significativa. El síntoma conductual más frecuente en los subtipos con afectación frontal fue la apatía (76%), no así en el subtipo temporal dominante. En el subgrupo de pacientes en los que se obtuvo muestra patológica, no se encontró una clara correlación entre el patrón anatómico y el subtipo patológico, excepto en los pacientes con EMN asociada y patología TDP, que presentaron atrofia restringida al lóbulo frontal. El grupo temporofrontoparietal, además de a patología TDP-43, se asoció en 3 sujetos a EA o DCB. Según estos autores, el subtipo frontotemporal podría ser un estadio evolutivo de la DFT vc, que se iniciaría como atrofia frontal dominante y se extendería posteriormente a los lóbulos temporales. El patrón temporofrontoparietal no se puede explicar de la misma forma, ya que la atrofia frontal encontrada en el mismo fue menor que en los de predominio frontal, y la atrofia temporal menor que la de las variantes con predominio temporal93.
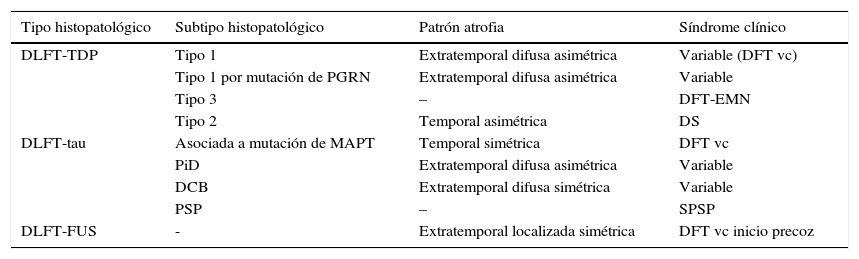
En 2011, Rohrer et al.94 presentaron un estudio retrospectivo en el que analizaron datos clínicos, neuropsicológicos y de neuroimagen (mediante volumetría y morfometría basada en vóxeles) de una cohorte de 95 casos con diagnóstico patológico de DLFT, y trataron de establecer un correlato clínico y anatómico para cada uno de los subtipos histopatológicos. Clasificaron los hallazgos anatómicos en 4 patrones basándose en la simetría o asimetría y la localización temporal o extratemporal de la atrofia. Observaron que existían determinados subtipos patológicos que se asociaban de forma significativa a determinados síndromes clínicos y que también existía asociación entre algunos subtipos y determinados patrones de atrofia (tabla 2).
Relación entre histopatología, patrón de atrofia y síndrome clínico
| Tipo histopatológico | Subtipo histopatológico | Patrón atrofia | Síndrome clínico |
|---|---|---|---|
| DLFT-TDP | Tipo 1 | Extratemporal difusa asimétrica | Variable (DFT vc) |
| Tipo 1 por mutación de PGRN | Extratemporal difusa asimétrica | Variable | |
| Tipo 3 | – | DFT-EMN | |
| Tipo 2 | Temporal asimétrica | DS | |
| DLFT-tau | Asociada a mutación de MAPT | Temporal simétrica | DFT vc |
| PiD | Extratemporal difusa asimétrica | Variable | |
| DCB | Extratemporal difusa simétrica | Variable | |
| PSP | – | SPSP | |
| DLFT-FUS | - | Extratemporal localizada simétrica | DFT vc inicio precoz |
Los subtipos DLFT-TDP 1-3 se han clasificado según el esquema de Mackenzie.
DCB: degeneración corticobasal; DFT-EMN: demencia frontotemporal asociada a enfermedad de la motoneurona; DFT vc: demencia frontotemporal variante conductual; DLFT-FUS: degeneración lobar frontotemporal asociada a la proteína de fusión en sarcoma; DLFT-TDP: degeneración lobar frontotemporal asociada a la proteína fijadora de ADN TAR 43; DLFT-tau: degeneración lobar frontotemporal asociada a tau; DS: demencia semántica; FUS: proteína de fusión en sarcoma; MAPT: microtubule associated protein; PGRN: progranulina; PiD: enfermedad de Pick; PSP: parálisis supranuclear progresiva; SCB: síndrome corticobasal; SPSP: síndrome de parálisis supranuclear progresiva.
Obtenido a partir de los datos de Rohrer et al.94.
Habitualmente, suele ser fácil distinguir las DFT de la EA desde un punto de vista clínico. No obstante, existen pacientes con DFT que comienzan con una alteración marcada de la memoria episódica y también pacientes con EA con presentación atípica, por ejemplo: variante de EA con alteración del lenguaje (generalmente APP logopénica), variante frontal de la EA (con alteraciones comportamentales/ejecutivas llamativas), atrofia cortical posterior (que habitualmente presenta alteración visuoespacial y/o visuoperceptiva, por lo que a veces recibe el nombre de «variante visual») o SCB asociado a EA.
La RM estructural, mediante morfometría basada en vóxeles, se ha mostrado útil para distinguir entre DFT y EA. En la DFT se observa atrofia de regiones frontales, insulares, cíngulo anterior y estriado frente a atrofia parietal posterior y occipital en la EA95. También es útil el análisis del grosor cortical regional, mostrando la EA mayor adelgazamiento cortical a nivel parietal y precuneal96.
Otras técnicas de RM útiles para distinguir la EA de la DLFT son la secuencia tensor de difusión97 (disminución de la anisotropía fraccional en regiones frontales en la DLFT), «arterial spin labelling»98,99 (hipoperfusión en regiones parietales y cíngulo posterior en la EA frente a hipoperfusión de lóbulos frontales en la DLFT) y técnicas combinadas de fluorodeoxiglucosa (FDG)-tomografía por emisión de positrones (PET)/RM100.
Diversos estudios realizados en variantes atípicas de la EA han mostrado cómo, de forma independiente a la presentación clínica, la EA se asocia a afectación del cíngulo posterior, precuneus, áreas parietales posteriores y temporales mediales101,102. Existen también estudios que compararon a pacientes con APP con y sin patología de EA. Se observó que aquellos con patología de EA (generalmente APP logopénica) presentaban mayor atrofia temporoparietal y los que tenían patología DLFT asociaban en la imagen un afilamiento característico del lóbulo temporal103,104.
Menos frecuentemente, las DLFT pueden confundirse con la demencia por cuerpos de Lewy (DCL), fundamentalmente en aquellos pacientes que presentan ilusiones o alucinaciones visuales105,106. Un estudio reciente de pequeño tamaño, ha sugerido que la escintigrafía con I123-MIBG puede ser útil para distinguir ambas entidades, mostrando la DCL una marcada reducción en la captación pero no la DLFT106. En otro estudio de pequeño tamaño, la RM no fue útil en el diagnóstico diferencial107.
Neuroimagen funcional: tomografía por emisión de positrones y tomografía computarizada de emisión de fotón único cerebralEn los últimos años, existe un uso creciente de las técnicas de neuroimagen funcional, como la Tc99m-hexametilpropileneamina oxima tomografía computarizada de emisión de fotón único (SPECT) o la F18-FDG-PET, en el diagnóstico de la DFT vc. En la DFT vc se observa una hipoperfusión/hipometabolismo en regiones frontales, mientras que la EA muestra una hipoperfusión temporoparietal y en cíngulo posterior108,109. Tanto la SPECT como la FDG-PET son más sensibles que la RM cerebral para detectar cambios precoces en la DFT vc110. La FDG-PET es también muy importante en la detección de fenocopias, que mostrarán un metabolismo conservado en regiones frontales. Sin embargo, en pacientes con clara atrofia cerebral en la RM, la FDG-PET no parece aportar mucho beneficio añadido.
Existen técnicas con PET que emplean nuevos radioisótopos. Una de ellas emplea un compuesto denominado compuesto C11-Pittsburgh B, que se une al β-amiloide y ha mostrado resultados prometedores en el diagnóstico diferencial entre EA y DFT111,112, especialmente en los casos con alteración del lenguaje113,114. Actualmente, se encuentran en desarrollo otras técnicas, como la PET de tau, la PET de PGRN o la PET de FUS, que se perfilan como herramientas diagnósticas prometedoras en un futuro a corto-medio plazo.
Biomarcadores en plasma y líquido cefalorraquídeoLos niveles en el líquido cefalorraquídeo del polipéptido de 42 aminoácidos de β-amiloide (A β42), que se encuentran disminuidos, y de tau, que se encuentran elevados, son marcadores sensibles y específicos para el diagnóstico de la EA115,116. El cociente o la ratio tau: A β42 es un parámetro útil para discriminar entre DLFT y EA, siendo menor en la DLFT117. En algunos pacientes con DFT-EMN, sin confirmación histopatológica, se han descrito niveles elevados de TDP-43 en el líquido cefalorraquídeo118.
Diversos estudios han mostrado que los pacientes con mutaciones en el gen de la PGRN presentan concentraciones plasmáticas de PGRN muy bajas54,55,119. La determinación de los niveles séricos de PGRN se ha propuesto como un método de cribado sensible y específico en estos pacientes120.
Se ha indicado también que los niveles plasmáticos de TDP-43 podrían estar relacionados con la afección cerebral en pacientes con DLFT121.
ConclusionesLa concepción de las DLFT desde un punto de vista molecular nos aproxima a un conocimiento más exacto de la enfermedad en términos de mecanismos causales y abre las puertas a nuevas opciones terapéuticas. Actualmente, se están desarrollando fármacos ligando-específicos, dirigidos contra dianas moleculares concretas.
La DFT vc es, de todas las variantes clínicas, la que presenta mayor heterogeneidad y en la que resulta más difícil predecir la enfermedad subyacente. Es posible que este síndrome sea un concepto demasiado amplio que en realidad englobe a entidades muy diferentes, por lo que la identificación de rasgos clínicos diferenciales y la definición de «subtipos clínicos» podría mejorar la correlación clinicopatológica. En cualquier caso, como complemento a la clínica, es necesario desarrollar herramientas que permitan realizar un diagnóstico patológico y molecular de la enfermedad in vivo.
La neuroimagen estructural y funcional, los biomarcadores analíticos y la genética se perfilan como herramientas útiles para establecer el diagnóstico, realizar el diagnóstico diferencial con otras entidades y predecir la enfermedad subyacente. De entre todos los biomarcadores mencionados, cabe destacar la importancia de la neuroimagen funcional por ser un instrumento más sensible que la neuroimagen estructural en fases tempranas y por el potencial de las técnicas con radiotrazadores específicos (11C-Pittsburgh B, tau, PGRN, FUS), que se perfilan como un instrumento con resultados prometedores en un futuro a corto plazo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

