



Las prionopatías representan hasta el 62% de los casos de demencia rápidamente progresiva (DRP) en los que se alcanza un diagnóstico definitivo. La variabilidad de los síntomas y signos iniciales y las diferencias en su evolución dificultan el diagnóstico precoz.
MétodosEstudio retrospectivo en el que se incluye a pacientes con prionopatía probable o definitiva, que acudieron a la consulta de Neurología de nuestro centro durante el periodo 1999-2012. Se describen las características clínicas y los resultados de las exploraciones complementarias (proteína 14-3-3, EEG, RM, PET-FDG y análisis genético), con la finalidad de identificar qué marcadores permiten un diagnóstico precoz.
ResultadosSe describe a 14 pacientes: 6 con enfermedad de Creutzfeldt-Jakob esporádica (ECJe) definitiva, 3 con ECJe probable, 4 con insomnio familiar fatal y uno con la nueva variante de la enfermedad de Creutzfeldt-Jakob. La mediana de edad al diagnóstico fue de 54 años y la mediana de supervivencia de 9,5 meses. El trastorno del ánimo fue el síntoma inicial más frecuente, seguido de inestabilidad de la marcha y deterioro cognitivo. La proteína 14-3-3 fue positiva en el líquido cefalorraquídeo en 7 de 11 pacientes y el EEG mostró signos típicos en 2 de 12 pacientes explorados. El estudio de neuroimagen mostró alteraciones en 13 de los 14 pacientes.
ConclusionesAdemás de la DRP, el trastorno conductual y de la marcha son síntomas iniciales frecuentes en las prionopatías. En nuestra serie, las pruebas complementarias más útiles para apoyar el diagnóstico fueron la RM y la PET-FDG.
Prionopathy is the cause of 62% of the rapidly progressive dementias (RPD) in which a definitive diagnosis is reached. The variability of symptoms and signs exhibited by the patients, as well as its different presentation, sometimes makes an early diagnosis difficult.
MethodsPatients withdiagnosis of definite or probable prionopathy during the period 1999-2012 at our hospital were retrospectively reviewed.The clinical features and the results of the complementary tests (14-3-3 protein, EEG, MRI, FDG-PET, and genetic analysis) were evaluated in order to identify some factors that may enable an earlier diagnosis to be made.
ResultsA total of 14 patients are described: 6 with definite sporadic Creutzfeldt-Jakob (sCJD) disease, 3 with probable sCJD, 4 with fatal familial insomnia, and 1 with the new variant. The median age at diagnosis was 54 years old. The mean survival was 9.5 months. Mood disorder was the most common feature, followed by instability and cognitive impairment. 14-3-3 protein content in the cerebrospinal fluid was positive in 7 of 11 patients, and the EEG showed typical signs in 2 of 12 patients. Neuroimaging (FDG-PET, MRI) studies suggested the diagnosis in 13 of the 14 patients included.
ConclusionsMost patients presenting with RPD suffer from a prion disease. In our series the most useful complementary tests were MRI and FDG-PET, being positive in 13 of the 14 patients studied.
Las prionopatías constituyen un conjunto de enfermedades neurodegenerativas producidas por el acúmulo de una isoforma anormal de la proteína priónica celular (PrPc) llamada PrPsc1. Se trata de una glucoproteína de la membrana plasmática compuesta por 209 aminoácidos y un puente disulfuro1, para la que la isoforma anormal tiene capacidad infecciosa en ausencia de ácidos nucleicos2. Está codificada por el gen PRNP del cromosoma 20, el cual presenta un polimorfismo de riesgo situado en el codón 129 para valina o metionina (M). La homocigosidad para M es un factor de riesgo para el desarrollo de esta afección. Las enfermedades priónicas se clasifican en adquiridas, esporádicas y hereditarias1. Su incidencia anual aproximada es de un caso por millón3. Una de las manifestaciones clínicas comunes a todas las prionopatías es el desarrollo de una demencia de rápida evolución. De hecho, hasta el 62% de los sujetos con una demencia rápidamente progresiva en los que se alcanza un diagnóstico definitivo4 tienen una enfermedad por priones. Sin embargo, la variabilidad de los síntomas y signos iniciales5 hace que solo en el 18% de los casos el diagnóstico sea preciso en la primera consulta6, pues normalmente se retrasa una media de 8 meses desde el inicio de los síntomas.
El objetivo de este trabajo es describir las características clínicas y la utilidad de determinadas exploraciones complementarias en el diagnóstico de enfermedad priónica. Para ello hemos realizado un estudio retrospectivo de los casos de prionopatía estudiados en la Clínica Universidad de Navarra.
Pacientes y métodosSe incluyó de forma retrospectiva a todos los pacientes con enfermedad priónica definitiva o probable, de acuerdo con los criterios diagnósticos validados7,8, estudiados en el Departamento de Neurología de la Clínica Universidad de Navarra entre los años 1999 y 2012. Los signos y síntomas clínicos, así como los resultados de las exploraciones complementarias, fueron recogidos mediante la revisión del historial médico. Dentro de las exploraciones complementarias, prestamos especial atención a la determinación de la proteína 14.3.3 en el LCR, el análisis del gen PRNP (estudio de mutaciones conocidas y valoración del polimorfismo en el codón 129) y los hallazgos electroencefalográficos, así como de neuroimagen estructural y funcional. En el caso de la RM, se analizó de forma visual la presencia o no de regiones hiperintensas, así como su localización, tanto en secuencias diffusion-weighted imaging (DWI) como fluid-attenuated inversion recovery (FLAIR). En los estudios de metabolismo cerebral mediante PET con 18-F-fluorodeoxiglucosa (PET-FDG) en 13 pacientes, y mediante SPECT con 99mTc- hexametil-propileno-aminaoxima (SPECT-HMPAO) en un paciente, se determinó visualmente la presencia o no de regiones de hipometabolismo y/o hipoperfusión y su distribución regional cerebral. En cada paciente, y a cada una de estas exploraciones, se le atribuyó un patrón de afectación en función de la existencia y localización de regiones hiperintensas, hipometabólicas y/o de hipoperfusión, respectivamente7–9. De esta forma, se establecen 3 patrones diferentes: un patrón cortical que implica la presencia de hiperintensidad, hipometabolismo y/o hipoperfusión en el córtex; el subcortical, caracterizado por la presencia de hiperintensidad, hipometabolismo y/o hipoperfusión en ganglios basales y/o tálamo, y, por último, el patrón córtico-subcortical, caracterizado por hiperintensidad, hipometabolismo y/o hipoperfusión en la región cortical y en las regiones subcorticales. Por otra parte, también se compararon los resultados obtenidos mediante RM y PET-FDG con la finalidad de analizar la sensibilidad de cada una de las técnicas. En todos los casos, se investigó si se había realizado examen neuropatológico y el resultado del mismo.
ResultadosCaracterísticas clínico-epidemiológicasSe identificó a 14 pacientes con enfermedades priónicas durante el periodo 1999-2012. Uno de ellos fue diagnosticado de ECJv confirmado mediante estudio anátomo-patológico7,10. En 4 pacientes se encontró la mutación D178N en el gen PrP asociada a IFF. Los 9 pacientes restantes se identificaron como ECJe, alcanzándose un diagnóstico de certeza en 6 de ellos8.
El 71,4% de los pacientes de nuestra serie fueron mujeres. La edad de inicio osciló entre los 27 y los 77 años, con una mediana de 54 años, siendo más jóvenes los casos de presentación familiar (Me = 44,5). La supervivencia del paciente con ECJv fue de 6 meses. La mediana de supervivencia en los casos con ECJe fue de 9,5 meses, mientras que en las formas familiares la supervivencia se prolongó una media de 20 meses.
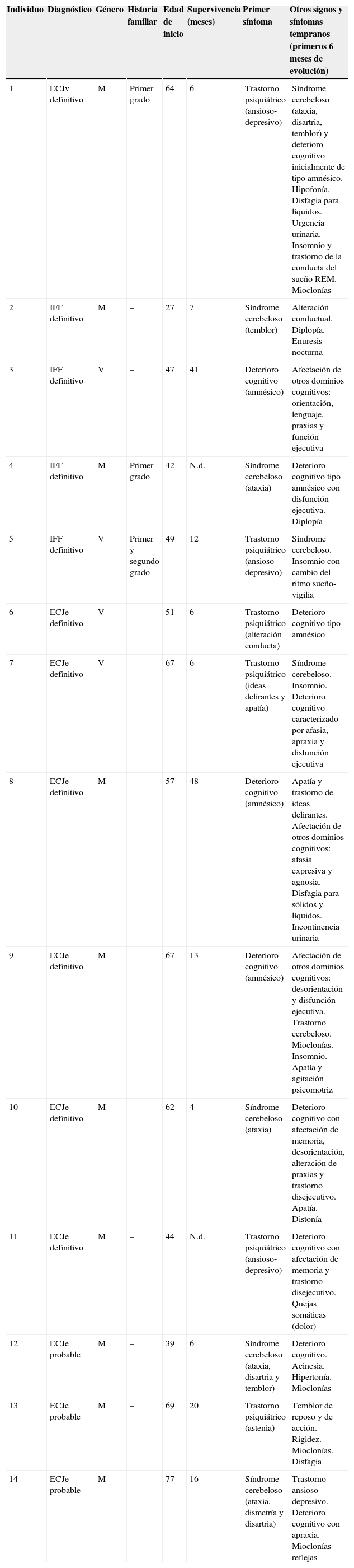
Un 42,85% de los pacientes de nuestra serie tuvo una afectación del estado de ánimo y de la conducta como primer síntoma clínico, caracterizado, en la mayoría de los casos, por un trastorno ansioso-depresivo con tendencia a la apatía, combinado con ideas delirantes. En un 35,71% de los casos, la enfermedad se presentó con un síndrome cerebeloso con ataxia de la marcha y temblor postural y cinético. En los 3 pacientes restantes, el motivo inicial de consulta fue un deterioro cognitivo aislado de rápida evolución. En la mayoría de los casos con deterioro cognitivo, la queja fundamental de los pacientes fueron problemas de memoria, si bien, tras realizar una evaluación neuropsicológica completa, se constató una afectación más extensa que englobaba otros dominios cognitivos, como orientación, lenguaje, praxias y función ejecutiva. No obstante, cabe resaltar que aunque los pacientes describían síntomas anteriormente referidos como iniciales, en todos ellos se apreció una combinación de los mismos en el momento de la exploración neurológica inicial. Otros síntomas y signos frecuentes durante los 6 primeros meses de evolución fueron las mioclonías (en 5 de los 14 pacientes), así como incontinencia urinaria, disfagia y rigidez muscular. Las características de los pacientes están resumidas en la tabla 1.
Características clínico-epidemiológicas
| Individuo | Diagnóstico | Género | Historia familiar | Edad de inicio | Supervivencia (meses) | Primer síntoma | Otros signos y síntomas tempranos (primeros 6 meses de evolución) |
|---|---|---|---|---|---|---|---|
| 1 | ECJv definitivo | M | Primer grado | 64 | 6 | Trastorno psiquiátrico (ansioso-depresivo) | Síndrome cerebeloso (ataxia, disartria, temblor) y deterioro cognitivo inicialmente de tipo amnésico. Hipofonía. Disfagia para líquidos. Urgencia urinaria. Insomnio y trastorno de la conducta del sueño REM. Mioclonías |
| 2 | IFF definitivo | M | – | 27 | 7 | Síndrome cerebeloso (temblor) | Alteración conductual. Diplopía. Enuresis nocturna |
| 3 | IFF definitivo | V | – | 47 | 41 | Deterioro cognitivo (amnésico) | Afectación de otros dominios cognitivos: orientación, lenguaje, praxias y función ejecutiva |
| 4 | IFF definitivo | M | Primer grado | 42 | N.d. | Síndrome cerebeloso (ataxia) | Deterioro cognitivo tipo amnésico con disfunción ejecutiva. Diplopía |
| 5 | IFF definitivo | V | Primer y segundo grado | 49 | 12 | Trastorno psiquiátrico (ansioso-depresivo) | Síndrome cerebeloso. Insomnio con cambio del ritmo sueño-vigilia |
| 6 | ECJe definitivo | V | – | 51 | 6 | Trastorno psiquiátrico (alteración conducta) | Deterioro cognitivo tipo amnésico |
| 7 | ECJe definitivo | V | – | 67 | 6 | Trastorno psiquiátrico (ideas delirantes y apatía) | Síndrome cerebeloso. Insomnio. Deterioro cognitivo caracterizado por afasia, apraxia y disfunción ejecutiva |
| 8 | ECJe definitivo | M | – | 57 | 48 | Deterioro cognitivo (amnésico) | Apatía y trastorno de ideas delirantes. Afectación de otros dominios cognitivos: afasia expresiva y agnosia. Disfagia para sólidos y líquidos. Incontinencia urinaria |
| 9 | ECJe definitivo | M | – | 67 | 13 | Deterioro cognitivo (amnésico) | Afectación de otros dominios cognitivos: desorientación y disfunción ejecutiva. Trastorno cerebeloso. Mioclonías. Insomnio. Apatía y agitación psicomotriz |
| 10 | ECJe definitivo | M | – | 62 | 4 | Síndrome cerebeloso (ataxia) | Deterioro cognitivo con afectación de memoria, desorientación, alteración de praxias y trastorno disejecutivo. Apatía. Distonía |
| 11 | ECJe definitivo | M | – | 44 | N.d. | Trastorno psiquiátrico (ansioso-depresivo) | Deterioro cognitivo con afectación de memoria y trastorno disejecutivo. Quejas somáticas (dolor) |
| 12 | ECJe probable | M | – | 39 | 6 | Síndrome cerebeloso (ataxia, disartria y temblor) | Deterioro cognitivo. Acinesia. Hipertonía. Mioclonías |
| 13 | ECJe probable | M | – | 69 | 20 | Trastorno psiquiátrico (astenia) | Temblor de reposo y de acción. Rigidez. Mioclonías. Disfagia |
| 14 | ECJe probable | M | – | 77 | 16 | Síndrome cerebeloso (ataxia, dismetría y disartria) | Trastorno ansioso-depresivo. Deterioro cognitivo con apraxia. Mioclonías reflejas |
En 11 de los 14 pacientes se analizó la presencia de la proteína 14-3-3 en el LCR11, siendo positiva en 7 casos (2 pacientes con IFF y 5 con ECJe) tras una media de evolución de 5,14 meses. En los 4 pacientes donde esta determinación fue negativa, la media de evolución fue similar (5,5 ± 2,51) en el momento de la valoración.
El EEG se realizó en 12 pacientes, siendo normal en un caso con IFF (a los 8 meses de evolución) y mostrando alteraciones inespecíficas en 9 pacientes (un enfermo con ECJv, 2 con diagnóstico de IFF y 6 con ECJe; media de seguimiento ± desviación estándar=4,89±2,71), consistentes en actividad de fondo mal diferenciada con ondas lentas en rango theta-delta polimorfas, difusas o localizadas, de carácter semicontinuo. Solamente en 2 pacientes con diagnóstico de ECJe se observó un patrón típico de enfermedad priónica, con presencia de descargas periódicas difusas bilaterales y de predominio anterior con patrón trifásico de intervalo corto (1Hz). En estos pacientes, el EEG se realizó a los 3 y 4 meses de evolución, respectivamente.
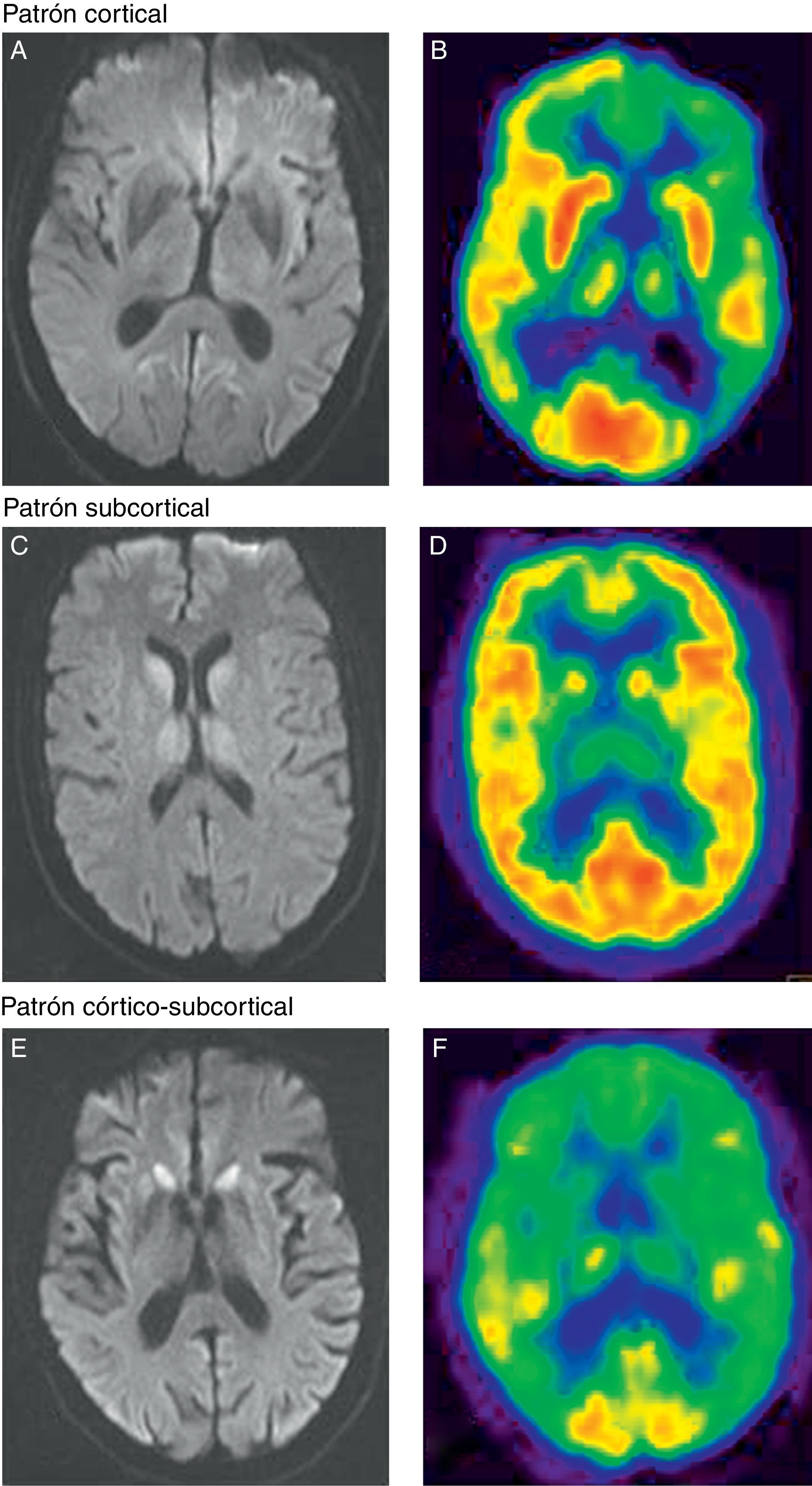
Con respecto a los resultados de neuroimagen, en los 13 pacientes en los que se realizó RM y/o PET-FDG se detectaron alteraciones indicativas de una enfermedad priónica (fig. 1). En todos los pacientes en los que se llevaron a cabo ambas pruebas, estas se realizaron en el mismo momento de la evolución. En 11 pacientes se realizó una RM cerebral con secuencias recomendadas, FLAIR y DWI, para el diagnóstico de ECJ. En la forma ECJv, aunque no llegó a apreciarse el típico signo del pulvinar12, destacaba una hiperintensidad talámica bilateral aislada10. En 2 pacientes no emparentados entre sí con IFF se realizó una RM. En uno se observó un patrón de afectación subcortical y en otro, hiperintensidad cortical, fundamentalmente en regiones límbicas y occipitales11.
 Hiperintensidad cortical fronto-parieto-occipital en el hemisferio izquierdo en secuencias de difusión. B) La PET-FDG muestra una disminución del metabolismo en la región cortical parietal, cingular posterior y frontal dorsolateral del hemisferio izquierdo y en los ganglios de la base izquierdos y ambos tálamos. Patrón subcortical. C) Hiperintensidad subcortical (ganglios basales y tálamo de forma bilateral) en secuencias de difusión. D) La PET-FDG muestra hipometabolismo cortical temporal izquierdo y subcortical (talámico bilateral). Patrón córtico-subcortical. E) Hiperintensidad en la región cortical frontal y parietal bilateral y simétrica, y subcortical (núcleo caudado y putamen) en secuencias de difusión. F) La PET-FDG muestra hipometabolismo cortical (corteza de asociación anterior y cingular) y subcortical (ganglios de la base).")
Patrón cortical. A) Hiperintensidad cortical fronto-parieto-occipital en el hemisferio izquierdo en secuencias de difusión. B) La PET-FDG muestra una disminución del metabolismo en la región cortical parietal, cingular posterior y frontal dorsolateral del hemisferio izquierdo y en los ganglios de la base izquierdos y ambos tálamos. Patrón subcortical. C) Hiperintensidad subcortical (ganglios basales y tálamo de forma bilateral) en secuencias de difusión. D) La PET-FDG muestra hipometabolismo cortical temporal izquierdo y subcortical (talámico bilateral). Patrón córtico-subcortical. E) Hiperintensidad en la región cortical frontal y parietal bilateral y simétrica, y subcortical (núcleo caudado y putamen) en secuencias de difusión. F) La PET-FDG muestra hipometabolismo cortical (corteza de asociación anterior y cingular) y subcortical (ganglios de la base).
En cuanto a los pacientes con ECJe, predominó un patrón de RM consistente en afectación córtico-subcortical en 4 de los 8 pacientes explorados con esta técnica. En 2 de los pacientes la afectación fue exclusivamente cortical y en otro exclusivamente subcortical (fig. 1).
Por otra parte, en 10 se realizó PET-FDG y en un paciente SPECT-HMPAO. En el paciente con ECJv, la PET-FDG mostró no solo un hipometabolismo talámico bilateral10, al igual que se apreciaba en la RM, sino también cortical, de predominio parietal y frontal izquierdo. De los 3 pacientes con IFF explorados con esta técnica, en 2 se obtuvo un patrón de afectación exclusivamente cortical y en el tercero, un hipometabolismo más extenso afectando también a regiones corticales13. En cuanto a los pacientes con ECJe, se realizó una PET-FDG en 7 y una SPECT-HMPAO en uno. En este grupo, predominó un patrón de afectación córtico-subcortical en 6 de 8 pacientes explorados. En los 2 pacientes restantes, destacaba la presencia de hipometabolismo a nivel subcortical.
En cuanto a la comparación de ambas técnicas de imagen, en 7 se constató una mayor sensibilidad de la PET-FDG sobre la RM, ya que en estos pacientes las regiones de hiperintensidad detectadas en RM fueron también observadas en el PET-FDG pero no al revés. Esta técnica permitió identificar otras regiones suplementarias de metabolismo cerebral alterado. En 2 pacientes el resultado fue el mismo con las 2 técnicas. Solamente en un caso el número de áreas afectadas detectadas fue mayor mediante la RM que mediante la PET-FDG.
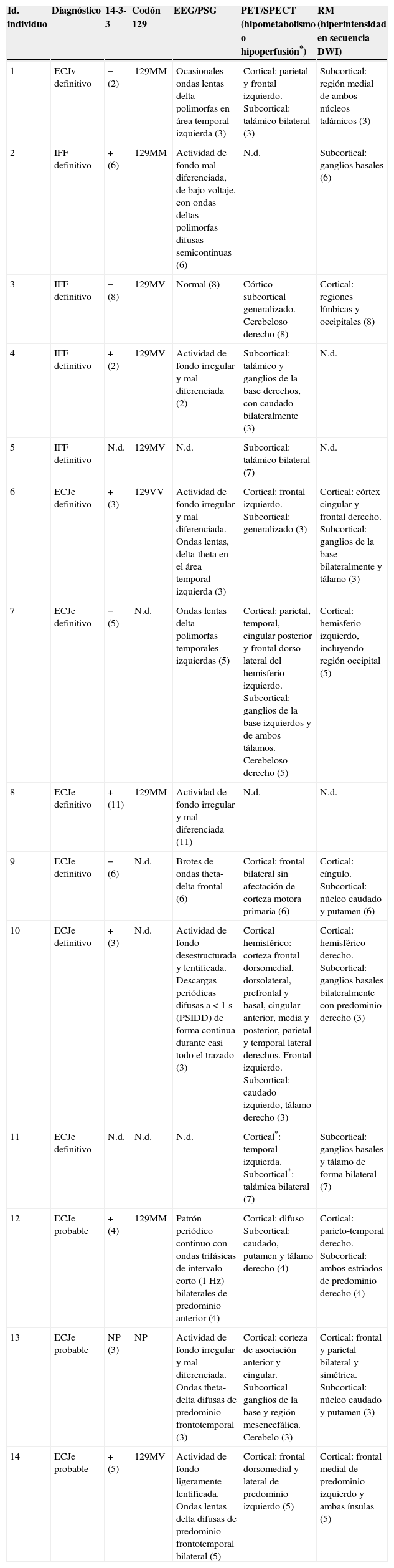
El estudio genético del gen PRNP se llevó a cabo en 9 pacientes. En 7 casos se realizó un estudio histológico post mortem. En la tabla 2 se resumen los hallazgos más significativos de las exploraciones complementarias realizadas en los pacientes.
Exploraciones complementarias
| Id. individuo | Diagnóstico | 14-3-3 | Codón 129 | EEG/PSG | PET/SPECT (hipometabolismo o hipoperfusión*) | RM (hiperintensidad en secuencia DWI) |
|---|---|---|---|---|---|---|
| 1 | ECJv definitivo | − (2) | 129MM | Ocasionales ondas lentas delta polimorfas en área temporal izquierda (3) | Cortical: parietal y frontal izquierdo. Subcortical: talámico bilateral (3) | Subcortical: región medial de ambos núcleos talámicos (3) |
| 2 | IFF definitivo | + (6) | 129MM | Actividad de fondo mal diferenciada, de bajo voltaje, con ondas deltas polimorfas difusas semicontinuas (6) | N.d. | Subcortical: ganglios basales (6) |
| 3 | IFF definitivo | − (8) | 129MV | Normal (8) | Córtico-subcortical generalizado. Cerebeloso derecho (8) | Cortical: regiones límbicas y occipitales (8) |
| 4 | IFF definitivo | + (2) | 129MV | Actividad de fondo irregular y mal diferenciada (2) | Subcortical: talámico y ganglios de la base derechos, con caudado bilateralmente (3) | N.d. |
| 5 | IFF definitivo | N.d. | 129MV | N.d. | Subcortical: talámico bilateral (7) | N.d. |
| 6 | ECJe definitivo | + (3) | 129VV | Actividad de fondo irregular y mal diferenciada. Ondas lentas, delta-theta en el área temporal izquierda (3) | Cortical: frontal izquierdo. Subcortical: generalizado (3) | Cortical: córtex cingular y frontal derecho. Subcortical: ganglios de la base bilateralmente y tálamo (3) |
| 7 | ECJe definitivo | − (5) | N.d. | Ondas lentas delta polimorfas temporales izquierdas (5) | Cortical: parietal, temporal, cingular posterior y frontal dorso-lateral del hemisferio izquierdo. Subcortical: ganglios de la base izquierdos y de ambos tálamos. Cerebeloso derecho (5) | Cortical: hemisferio izquierdo, incluyendo región occipital (5) |
| 8 | ECJe definitivo | + (11) | 129MM | Actividad de fondo irregular y mal diferenciada (11) | N.d. | N.d. |
| 9 | ECJe definitivo | − (6) | N.d. | Brotes de ondas theta-delta frontal (6) | Cortical: frontal bilateral sin afectación de corteza motora primaria (6) | Cortical: cíngulo. Subcortical: núcleo caudado y putamen (6) |
| 10 | ECJe definitivo | + (3) | N.d. | Actividad de fondo desestructurada y lentificada. Descargas periódicas difusas a < 1 s (PSIDD) de forma continua durante casi todo el trazado (3) | Cortical hemisférico: corteza frontal dorsomedial, dorsolateral, prefrontal y basal, cingular anterior, media y posterior, parietal y temporal lateral derechos. Frontal izquierdo. Subcortical: caudado izquierdo, tálamo derecho (3) | Cortical: hemisférico derecho. Subcortical: ganglios basales bilateralmente con predominio derecho (3) |
| 11 | ECJe definitivo | N.d. | N.d. | N.d. | Cortical*: temporal izquierda. Subcortical*: talámica bilateral (7) | Subcortical: ganglios basales y tálamo de forma bilateral (7) |
| 12 | ECJe probable | + (4) | 129MM | Patrón periódico continuo con ondas trifásicas de intervalo corto (1 Hz) bilaterales de predominio anterior (4) | Cortical: difuso Subcortical: caudado, putamen y tálamo derecho (4) | Cortical: parieto-temporal derecho. Subcortical: ambos estriados de predominio derecho (4) |
| 13 | ECJe probable | NP (3) | NP | Actividad de fondo irregular y mal diferenciada. Ondas theta-delta difusas de predominio frontotemporal (3) | Cortical: corteza de asociación anterior y cingular. Subcortical ganglios de la base y región mesencefálica. Cerebelo (3) | Cortical: frontal y parietal bilateral y simétrica. Subcortical: núcleo caudado y putamen (3) |
| 14 | ECJe probable | + (5) | 129MV | Actividad de fondo ligeramente lentificada. Ondas lentas delta difusas de predominio frontotemporal bilateral (5) | Cortical: frontal dorsomedial y lateral de predominio izquierdo (5) | Cortical: frontal medial de predominio izquierdo y ambas ínsulas (5) |
Entre paréntesis se indican los meses de evolución de la enfermedad en el momento de la realización de cada una de las exploraciones complementarias.
N.d.: no disponible.
* En el paciente número 11 se demostró hipoperfusión cortical y subcortical mediante SPECT.
En este artículo, presentamos una descripción de 14 pacientes con distintas formas de prionopatía procedentes de diversas regiones de la geografía española (Navarra, País Vasco, Madrid, Andalucía, Valencia, Castilla y León, y La Rioja), estudiados en nuestro centro entre los años 1999 y 2012.
En nuestra serie, los casos de ECJe fueron más frecuentes que los IFF y ECJv, si bien estas formas de enfermedad priónica representan un alto porcentaje en nuestra serie debido a un sesgo de referencia. Únicamente en uno de ellos la enfermedad comenzó antes de los 50 años, lo que coincide con la edad de presentación descrita para esta enfermedad14. Respecto a la variante familiar, todos los casos eran portadores de la mutación D178N, la cual ya ha sido descrita previamente como una mutación frecuente en familias del País Vasco con prionopatía15. En el caso del paciente con ECJv, el comienzo clínico fue más tardío que el habitual7 y el tiempo de supervivencia menor.
En nuestros pacientes, el síntoma inicial más frecuente fue una afectación del estado de ánimo y la conducta, mientras que otros estudios establecen como síntoma de comienzo de la enfermedad el deterioro cognitivo5 o un síndrome cerebeloso5,16. Aunque este dato varía según las series, y puede ser el responsable del retraso en el diagnóstico5, todos los autores coinciden en la alta proporción de pacientes en los que se combinan 3 tipos de síntomas: deterioro cognitivo, trastornos psiquiátricos de tipo ansioso-depresivo y síndrome cerebeloso. Apoyándonos tanto en nuestros datos como en los publicados en la literatura, creemos que la presencia de esta tríada sintomática podría ser considerada como un criterio de alta sospecha de prionopatía.
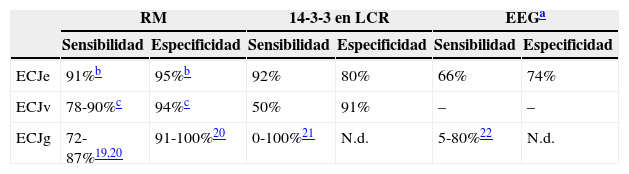
En el diagnóstico de estas enfermedades resulta de gran utilidad las exploraciones complementarias. La presencia de la proteína 14-3-3 en el LCR es un marcador de daño neuronal que indica el diagnóstico de prionopatía17. Estudios previos han demostrado que su detección en LCR es sensible y específica para el diagnóstico de ECJe, habiendo sido incluida en el año 2009 como uno de los criterios que apoyan el diagnóstico de la enfermedad8. Recientemente, Muayqil et al.18, en una revisión de pacientes con diagnóstico definitivo o probable de ECJe, atribuyen a la presencia de la proteína 14-3-3 en el LCR unos valores de sensibilidad del 92% y una especificidad del 80% (tabla 3). Cuando además existen cambios típicos en el EEG (complejos de ondas agudas periódicas)8, adquieren un elevado valor predictivo positivo23, mientras que si no existen ambos parámetros se recomienda considerar otros diagnósticos alternativos23. No obstante, estas determinaciones también tienen falsos positivos, habiéndose descrito pacientes con diagnóstico de enfermedad priónica en los que el estudio anatomopatológico fue compatible con enfermedad de Alzheimer o demencia por cuerpos de Lewy24. En 2 de nuestros pacientes con diagnóstico definitivo de ECJe no se detectaron cambios típicos en el EEG y la determinación de la proteína 14-3-3 resultó negativa. Estos datos indican que no debería excluirse el diagnóstico de prionopatía en los casos que no existan resultados positivos en el EEG o el LCR si persiste la sospecha clínica y se han descartado otras posibles causas. Por otra parte, en el caso de la enfermedad familiar por priones, el valor diagnóstico de la proteína 14-3-3 en el LCR es menor23, aunque en nuestra serie esta determinación fue positiva en 2 de los 3 pacientes explorados.
Sensibilidad y especificidad de las exploraciones complementarias
| RM | 14-3-3 en LCR | EEGa | ||||
|---|---|---|---|---|---|---|
| Sensibilidad | Especificidad | Sensibilidad | Especificidad | Sensibilidad | Especificidad | |
| ECJe | 91%b | 95%b | 92% | 80% | 66% | 74% |
| ECJv | 78-90%c | 94%c | 50% | 91% | – | – |
| ECJg | 72-87%19,20 | 91-100%20 | 0-100%21 | N.d. | 5-80%22 | N.d. |
ECJe: enfermedad de Creutzfeldt-Jakob esporádica; ECJg: enfermedad de Creutzfeldt-Jakob genética; ECJv: nueva variante de la enfermedad de Creutzfeldt-Jakob; N.d.: no disponible.
En los últimos años, los estudios de neuroimagen han cobrado especial relevancia en el diagnóstico de las prionopatías25, habiendo sido incluidos los hallazgos de RM cerebral en los criterios diagnósticos, tanto de ECJv7 como en la ECJe8. La presencia del «signo del pulvinar» en la RM cerebral es un dato que apoya el diagnóstico de ECJv12, con una sensibilidad de entre el 78 y el 90% (tabla 3). Este signo consiste en hiperintensidad simétrica en pulvinar bilateral con respecto a la corteza cerebral y la parte anterior del putamen. Es importante una correcta identificación de este hallazgo radiológico, dado que en la ECJe puede hallarse un falso signo del pulvinar en el que la hiperintensidad en este núcleo es inferior a la detectada en putamen y caudado9. En la ECJe, la utilización conjunta de las secuencias FLAIR y DWI de la RM cerebral tiene mayor capacidad de detección de la enfermedad26,27 que la proteína 14-3-3 y el EEG, con una sensibilidad diagnóstica del 91% y una especificidad del 95%28. De los 9 pacientes con ECJe descritos en nuestra serie, disponemos del estudio de imagen de 7, habiendo encontrado hallazgos característicos en todos ellos (alteración de señal en núcleos putamen y caudado o en al menos 2 regiones corticales)8. En estos 7 pacientes también se realizó un estudio PET-FDG, que puso de manifiesto un hipometabolismo córtico-subcortical. Respecto a las prionopatías familiares, aunque se han descrito cambios inespecíficos consistentes en dilatación ventricular y atrofia de la corteza cerebral y cerebelosa, no existe en la actualidad un patrón específico de RM cerebral. En 3 de los pacientes con prionopatía familiar se completó el estudio de metabolismo cerebral y se objetivó un extenso hipometabolismo de predominio en el tálamo, siendo los resultados congruentes con la literatura29,30. De acuerdo con estas observaciones, el hipometabolismo córtico-subcortical en el PET-FDG podría ser más característico de las formas esporádicas, mientras que en las familiares este sería fundamentalmente talámico.
Los datos descritos de nuestra serie apoyan la importancia de las técnicas de neuroimagen, dado que las pruebas de RM y/o PET-FDG orientaron el diagnóstico en los 13 casos en los que se llevaron a cabo, y en 5 pacientes con diagnóstico de certeza fueron la única prueba positiva.
En resumen, la combinación de síntomas psiquiátricos, cognitivos y cerebelosos es altamente indicativa de enfermedad priónica. En cuanto a las exploraciones complementarias, los hallazgos típicos en las pruebas de imagen (RM y/o PET-FDG) orientan el diagnóstico de prionopatía con una alta sensibilidad. En nuestra serie, la PET-FDG permitió identificar regiones cerebrales suplementarias de metabolismo alterado, indicando una mayor capacidad diagnóstica que la RM, de acuerdo con otras publicaciones recientes31,32.
Por tanto, ante la sospecha clínica de prionopatía, es fundamental incluir en el proceso de estudio RM cerebral con secuencias DWI y FLAIR y/o estudio PET-FDG. Los datos de nuestra serie indican que, en un futuro, la PET-FDG podría tener un papel en el diagnóstico de esta enfermedad, aunque en la actualidad esta exploración no está incluida en los criterios diagnósticos vigentes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Este artículo fue presentado como Comunicación Oral en la Reunión Anual de la Sociedad Española de Neurología del año 2012.

