



Las malformaciones cavernosas cerebrales (CCM; OMIM 116860) son engrosamientos cavernosos vasculares sin intervención del parénquima cerebral con una prevalencia estimada en la población general del 0,1-0,5%. La cavernomatosis cerebral presenta un patrón de herencia autosómico dominante con penetrancia clínica y radiológica incompleta. Tres genes se han asociado al desarrollo de lesiones: CCM1/KRIT1, CCM2/MGC4607 y CCM3/PDCD10.
DesarrolloLa mutación responsable no es detectada en un alto porcentaje de casos, por lo que nuevos enfoques son necesarios para su detección. En esta revisión se analizan las técnicas actualmente utilizadas y las posibles mutaciones o variantes que pueden ser detectadas en un laboratorio de genética molecular o biología molecular. Asimismo, se analizan alternativas que pueden ser abarcadas para la detección de mutaciones en aquellos pacientes en los que los estudios hayan resultado negativos.
ConclusionesEl diagnóstico molecular de la cavernomatosis cerebral debe incluir al menos la detección del número de copias y la secuenciación de los genes CCM. Finalmente, ofrecer un adecuado consejo genético es crucial para proporcionar información y apoyo a los pacientes y familias que padecen la enfermedad.
Cerebral cavernous malformations (CCMs; OMIM 116860) are enlarged vascular cavities without intervening brain parenchyma whose estimated prevalence in the general population is between 0.1% and 0.5%. Familial CCM is an autosomal dominant disease with incomplete clinical and radiological penetrance. Three genes have been linked to development of the lesions: CCM1/KRIT1, CCM2/MGC4607, and CCM3/PDCD10.
Development: The aetiological mutation is not detected in a large percentage of cases and new approaches are therefore needed. The aim of this review is to analyse current molecular techniques and the possible mutations or variations which can be detected in a molecular genetics or molecular biology laboratory. Likewise, we will analyse other alternatives that may help detect mutations in those patients showing negative results.
ConclusionsA molecular diagnosis of cerebral cavernous malformations should provide at least the copy number variation and sequencing of CCM genes. In addition, appropriate genetic counselling is a crucial source of information and support for patients and their relatives.
Las malformaciones vasculares cerebrales son lesiones relativamente comunes que representan un grupo heterogéneo de trastornos con una historia natural e implicaciones clínicas muy distintas. Estas lesiones pueden causar desde afecciones totalmente benignas, hasta serias discapacidades neurológicas e incluso la muerte en una proporción significante de individuos. Las más comunes son las malformaciones arteriovenosas (AVM) y las malformaciones cavernosas cerebrales (CCM; OMIM #116860), que afectan de forma casi exclusiva al sistema nervioso central. Los síntomas asociados a estas malformaciones vasculares incluyen: infartos hemorrágicos debido a la rotura de las lesiones, epilepsia y signos neurológicos focales.
Las CCM son lesiones de tipo venoso y no visibles mediante arteriografía, de ahí que fueran referidas como angiográficamente negativas debido a su flujo lento. Existen múltiples términos que se han empleado indistintamente para denominar estas lesiones, como son: angioma cavernoso, cavernoma, hemangioma cavernoso y malformación venosa cavernosa. Su prevalencia se estima entre el 0,1 y el 0,5%1,2 y se crean por la dilatación de agrupaciones de vasos sanguíneos bien delimitados sin intervención del parénquima3.
Las lesiones pueden ocurrir tanto de forma esporádica como de forma hereditaria. A nivel familiar, la cavernomatosis cerebral presenta un patrón de herencia autosómico dominante con penetrancia clínica y radiológica incompleta. Mientras que algunos cavernomas son clínicamente silentes, otros pueden llegar a causar convulsiones, hemorragias o déficits neurológicos focales. Las lesiones cavernomatosas representan entre el 5 y el 15% de todas las malformaciones vasculares del sistema nervioso central. Los cavernomas pueden ser asintomáticos y ser encontrados de manera casual en el curso de una exploración mediante resonancia magnética. Sin embargo, el 50-70% llegan a manifestarse clínicamente entre la segunda y cuarta décadas, sin distinción de sexos. También se han descrito casos de cavernomatosis cerebral en la infancia4. La incidencia real de los cavernomas es difícil de estimar, debido a que pueden ser confundidos con otros tipos de malformaciones vasculares3, y solo existen datos aproximados de lo que acontece en la población general a través de estudios de autopsias y de resonancia magnética. En 1991, 2 estudios5,6 analizaron una gran serie de resonancias magnéticas, estimando una prevalencia del 0,39 y del 0,47%. Estudios posteriores confirman estos datos, estableciéndose la prevalencia de la enfermedad en la población general entre el 0,1 y el 0,5%1,2.
Los cavernomas pueden dar cuadros de alteraciones neurológicas locales, con crisis de epilepsia, o generales, tipo cefaleas y accidentes cerebrovasculares con hemorragia. De hecho, la hemorragia intracraneal es la complicación más temida, ya que puede provocar una grave discapacidad o, incluso, la muerte7. Actualmente, la detección de las lesiones de CCM se realiza mediante imagen por resonancia magnética (MRI) en eco-gradiente en T2, debido a que clínicamente pueden compartir sintomatología con otras malformaciones vasculares o con microhemorragias cerebrales de distinta etiología. Los pacientes de cavernomatosis cerebral se pueden clasificar en 2 grupos:
- 1.
Forma familiar. Se considera así cuando hay más de un enfermo en la familia o un solo afectado con múltiples lesiones (cavernomatosis múltiple).
- 2.
Forma esporádica. Se considera como tal cuando se trata de un único enfermo y con un solo cavernoma en MRI con eco-gradiente.
Existen estudios que apuntan a la posibilidad de que un 75% de los casos esporádicos sean en realidad formas familiares, con lesiones asintomáticas que enmascaran el patrón de segregación autosómico dominante8.
DesarrolloDiagnóstico genéticoActualmente la detección de mutaciones en pacientes CCM no es completa: desde un 78%9 hasta un 40%10,11 en los estudios más recientes, por lo que el abordaje molecular no puede ser empleado para descartar la enfermedad. Estas diferencias pueden deberse a la inclusión de pacientes con historia de hemorragia cerebral de etiología desconocida. De hecho, la Sociedad Española de Neurología recomienda el estudio genético de cavernomatosis en el diagnóstico del ictus12.
Tres genes se han asociado a la cavernomatosis cerebral: CCM1/KRIT1, situado en el brazo largo del cromosoma 7 (7q21-q22)13,14; CCM2/MGC4607, brazo corto del cromosoma 7 (7p13)15, y CCM3/PDCD10, localizado en el cromosoma 3 (3q26.1)16. Es importante resaltar que cerca de 200 mutaciones diferentes han sido descritas en pacientes CCM con un bajo grado de recurrencia. Sin embargo, en el caso de la península Ibérica se ha observado cierta prevalencia de una deleción de 14 pares de bases (pb) en el exón 5 de CCM217. Por otro lado, la mutación c.1363T> C en CCM1, altamente prevalente en población hispana14, no fue detectada en pacientes españoles18. En población ibérica CCM1 es el responsable del 56% de las mutaciones encontradas en formas familiares, mientras que CCM2 lo es del 33% de ellas y CCM3 solo del 6%10. Existe aún un alto porcentaje de familias en las que no ha sido detectada la mutación responsable. Por otro lado, grandes deleciones han sido detectadas en pacientes con cavernomatosis en los 3 genes CCM11,19-21.
Los 3 genes codifican proteínas de andamiaje implicadas en diferentes procesos, incluyendo angiogénesis y vasculogénesis, entre otros. Las 3 proteínas interaccionan entre sí formando un complejo ternario. Por ello, mutaciones o variantes que produzcan una pérdida de la funcionalidad de las proteínas son consideradas como patogénicas. Esto no es siempre sencillo de dilucidar. Podemos encontrarnos con las siguientes posibilidades: variantes nonsense, frameshift, en zona de splicing, missense, silentes y grandes deleciones.
Variantes de tipo nonsense y frameshiftLas variantes nonsense son las que introducen un codón de parada prematuro, por ejemplo c.902C>G (p.S301X) en CCM1, que produce una proteína truncada de 301 aminoácidos (aa) en lugar de la proteína completa de 736aa. Las variantes frameshift son aquellas en las que se observan inserciones, deleciones o, también, indels (inserciones y deleciones en la misma región), que llevan a la introducción de codones de parada prematuros. Como ejemplo en CCM1, la inserción c.968_971dupCACC (p.Ile325ThrfsX11) provoca la producción de una proteína truncada de 336aa. Este tipo de variantes son patogénicas, siendo las que encontramos con mayor frecuencia10,11.
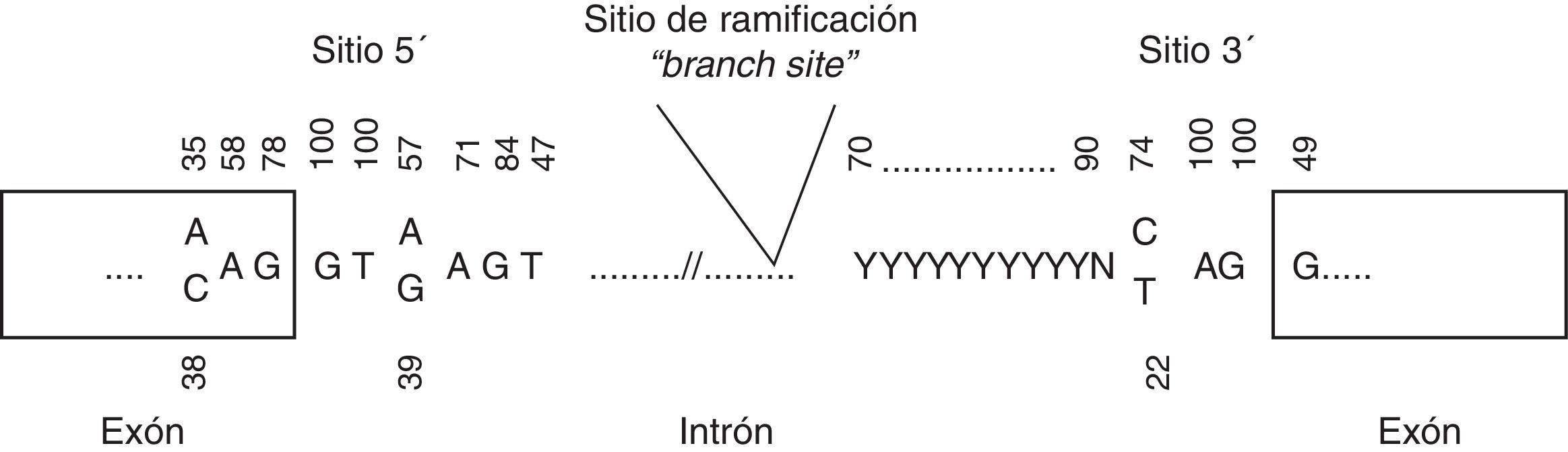
Variantes en zonas de splicingPodemos diferenciar 2 tipos de variantes en esta región: a)en la región invariante de splicing, definidas por las 2 bases intrónicas adyacentes al sitio de splicing, y b)en la región variante de splicing, definidas por las bases 1-3 dentro del exón y 3-8 del intrón respecto al sitio de splicing (fig. 1). Las variantes en la región invariante alteran siempre el splicing, pudiendo provocar la eliminación del exón, o parte del exón, o retención completa o parcial del intrón en el transcrito maduro. Esto, a su vez, conlleva la producción de una proteína truncada o proteína más corta, dependiendo de la modificación o no de la pauta de lectura. Por ello, son consideradas como patogénicas. En estos casos sería recomendable realizar un estudio a nivel de ADNc para conocer las consecuencias exactas.
 y aceptor (3′) de splicing y punto de ramificación. Los números indican las frecuencias de cada nucleótido en cada posición.")
Por otro lado, las variantes en la región variante de splicing pueden ser o no patogénicas, y por esto son consideradas como variantes de significado incierto. Para definir su patogenicidad sería imprescindible estudiar el ADNc. Con este tipo de estudios podemos valorar la producción de splicing crípticos, es decir, la elección de un sitio de splicing distinto al silvestre que lleve a la producción de una proteína truncada. Podemos valorar in silico la producción de splicing crípticos mediante herramientas como Human Splicing Finder (http://www.umd.be/HSF/)22.
Variantes missense y silentesLas variantes de tipo missense son las que producen un cambio de aminoácido, manteniéndose sin cambios a nivel proteico en el caso de las variantes silentes. Ambos casos deben de considerarse como variantes de significado incierto, por lo que sería recomendable realizar un estudio a nivel de ADNc para valorar la producción de splicing crípticos. Hay diversos casos publicados tanto de variantes missense en CCM que activan sitios crípticos de splicing10,23-25 como de variantes silentes15,26,27. Otro mecanismo por el que estas variantes pueden ser patogénicas sería a través de la producción de una variante aberrante sujeta a degradación, donde solo se observaría el alelo normal en ADNc26. En el caso de observar ambos alelos en ADNc, las variantes silentes serán definidas como no patogénicas10,11. Sin embargo, no podríamos decir lo mismo de las variantes missense, que mantendrían su condición de patogenicidad incierta. Existen aproximaciones bioinformáticas, como PolyPhpen-2 y SIFT, que utilizan algoritmos para predecir el efecto de la variante en la secuencia proteica en base a datos de estructura y secuencia de la proteína28,29. No obstante, harían falta estudios proteómicos para la confirmación de la condición patogénica.
Grandes delecionesLa introducción de la técnica Multiplex Ligation-dependent Probe Amplification (MLPA) (P130 y P131, MRC-Holland, Países Bajos), basado en la amplificación dependiente de ligación de varias sondas al mismo tiempo, ha permitido la detección de casos en los que hay deleciones superiores a un exón, algo que no es infrecuente. Asimismo, el método Quantitative Multiplex PCR of Short Fluorescent fragments (QMPSF) ha sido recientemente utilizado para la detección de deleciones11. Hasta ahora se han detectado cerca de 40 deleciones en los genes CCM10,11,16,20,21,27,30. Hay que decir que estas deleciones no son detectables mediante secuenciación directa y han de considerarse como patogénicas, puesto que suelen producir la ausencia del transcrito completo.
Polimorfismos y variantes intrónicasEn el transcurso de la secuenciación podemos encontrarnos con diversos polimorfismos que pueden presentarse tanto en región exónica, missense o silentes, como en región intrónica. Frente a estas variantes debemos primero consultar bases de datos como la de los 1.000 genomas (http://browser.1000genomes.org/index.html). Algunos de estos polimorfismos se han asociado con un aumento en el riesgo de sufrir CCM, mientras que otros predisponen a una mayor ocurrencia de una sintomatología «potencialmente discapacitante» (como jaquecas) más que a una sintomatología con posible riesgo para la salud» (como epilepsia o hemorragia cerebral)24.
En el caso de variantes intrónicas fuera de los sitios de splicing y que no son polimorfismos, debería realizarse un estudio a nivel de ADNc para valorar la producción de splicing crípticos. Hay casos descritos, como c.1564-14C>G y c.2026-12A>G en CCM1, que provocan la eliminación de exón 12 y 15, respetivamente, produciendo una proteína truncada en el primer caso y una proteína más corta en el segundo11.
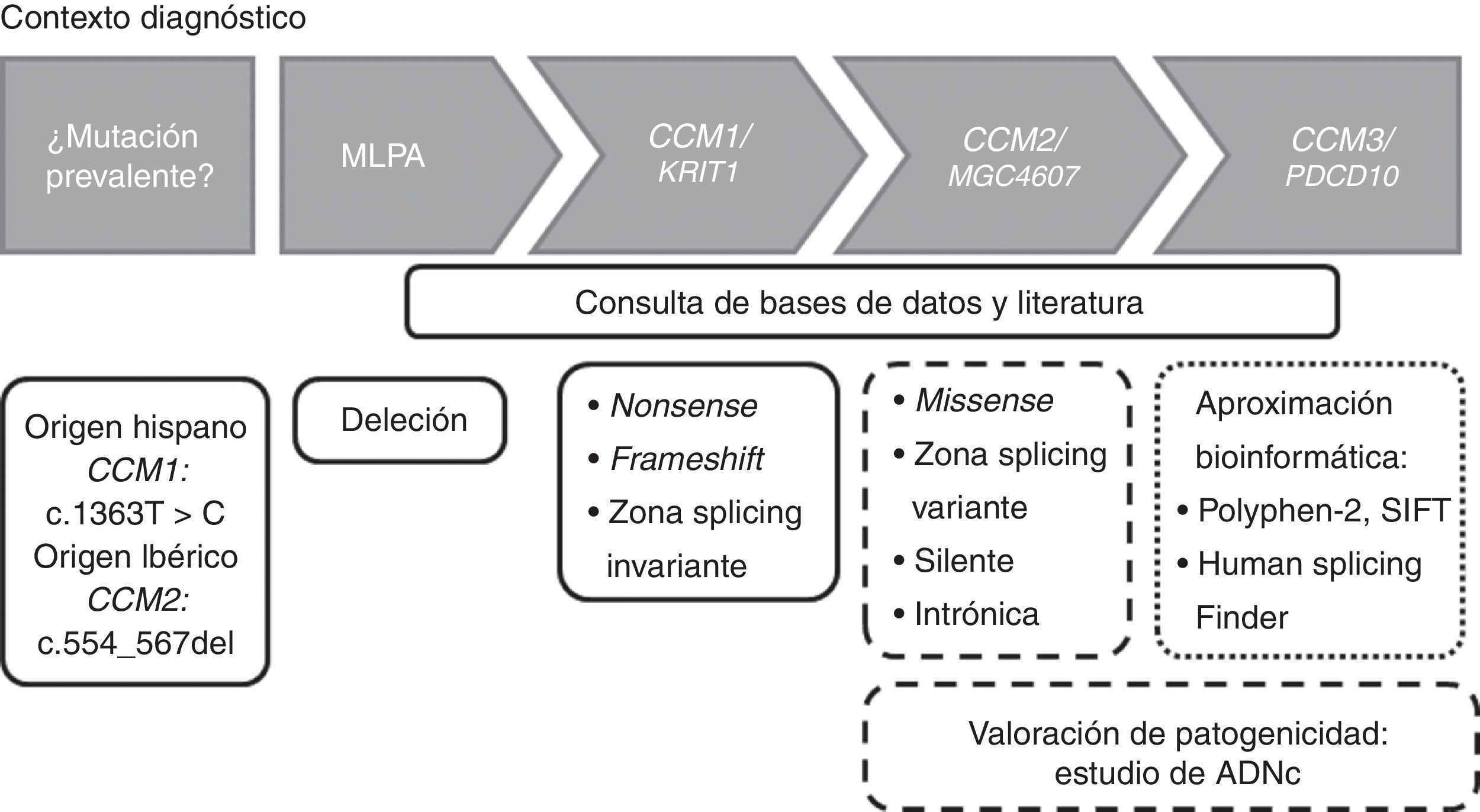
Estrategia de genotipadoLa estrategia de genotipado puede variar dependiendo del origen étnico del sujeto de estudio. Como hemos comentado antes, dependiendo el origen étnico puede realizarse en un primer momento la detección de la mutación c.1363T>C en CCM1 en población hispana, o la deleción de 14pb en el exón 5 en CCM2 (c.554_567del) en población ibérica. En general, y basándonos en nuestra experiencia, para el estudio molecular de la cavernomatosis proponemos el diagrama de flujo de la figura 2.

Diagrama de flujo para la detección de mutaciones en los genes CCM en un contexto diagnóstico. Los recuadros con línea continua indican que dichas mutaciones son patogénicas. Los recuadros con líneas discontinuas indican que ese tipo de mutaciones deberían ser estudiadas a nivel de ADNc para valorar la patogenicidad.
En un primer momento realizamos una búsqueda de deleciones en los 3 genes CCM mediante kits de MLPA (P130 y P131, MRC-Holland, Países Bajos). Si el número de copias es normal, procedemos entonces a secuenciar mediante el método Sanger y de forma seriada las regiones exónicas e intrónicas adyacentes de los genes CCM. Esto es, secuenciamos primero el gen CCM1, después CCM2 y finalmente CCM3. Si detectáramos variantes que introducen un codón de parada prematuro (nonsense o frameshift) o localizadas en zona de splicing invariante daríamos por finalizado el estudio, debido a que estas mutaciones producen pérdida de funcionalidad y son por ello patogénicas. En el caso de encontrar una variante missense, una transición silente o variante intrónica, seguiríamos secuenciando debido a su significado incierto. En estos casos, junto con variantes localizadas en la región de splicing variante sería altamente recomendable un análisis a nivel de ADNc para valorar la producción de splicing crípticos.
Perspectivas futuras/Otras alternativasDebido a que la mutación responsable de la enfermedad puede no ser hallada en un porcentaje alto de pacientes, se podría abordar el diagnóstico genético mediante otras alternativas.
- i)
Estudios de los tres genes CCM a nivel de ADNc. A través de esta alternativa, Riant et al.31 determinaron la inclusión de 99pb a la secuencia nucleotídica del transcrito completo de CCM1, correspondiente al intrón 2. En posterior análisis en ADN detectaron la deleción c.262+132_262+133delAA, que provoca la activación de un sito de splicing adyacente. La inclusión de dicho fragmento modifica el marco de lectura, introduciendo un codón de parada y produciendo, por tanto, una proteína truncada. Hay que recalcar que esta mutación no se habría detectado mediante el diagrama de flujo seguido en la figura 2.
- ii)
Las regiones 5′ y 3′UTR no son estudiadas por lo general. Estas regiones tienen bastante influencia en la estabilidad del ARNm32,33. La región 5′UTR es especialmente compleja en CCM1, pudiendo transcribir de forma diferencial hasta 30 variantes con diferentes combinaciones de exones no codificantes. Recientemente se ha informado de 2 mutaciones contiguas en la región 3′UTR de CCM1 en un mismo paciente que podría provocar la interacción con un miRNA24, lo que podría llevar a una menor expresión de Krit1. Por ello, mutaciones en estas regiones podrían alterar la estabilidad del ARNm y disminuir la producción de proteína funcional. Sin embargo, la confirmación de la condición patogénica no es sencilla debido a la dificultad de obtener células endoteliales de lesiones de pacientes CCM.
- iii)
Actualmente, las nuevas técnicas de secuenciación masiva (Next Generation Sequencing [NGS]) se han abierto paso en el ámbito privado, aunque con una lenta incorporación en los laboratorios públicos. Estas nuevas técnicas aumentan el rendimiento y la velocidad de generación de los datos, con una drástica reducción de costes de secuenciación por base. Con esta técnica se puede analizar de forma simultánea los 3 genes CCM. Una alternativa interesante sería analizar tanto ADN como cDNA en la misma carrera. Hasta la fecha no hay datos con esta tecnología para la detección de mutaciones en la cavernomatosis. Por ello, cada laboratorio deberá establecer el coste-beneficio de la tecnología usada en su diagrama de flujo. La técnica de análisis High Resolution Melt34 puede disminuir el coste global secuenciando solo aquellos fragmentos en los que se detecte una variación en la curva de disociación, algo que habría que tener en cuenta.
- iv)
Desde hace años se especula que un cuarto gen podría estar implicado35. En pacientes con historia familiar de cavernomatosis, en los que los estudios genéticos han resultado negativos, podría llevarse a cabo un estudio de exoma completo mediante NGS. Las variantes identificadas aquí deberán ser validadas clínicamente para evitar una avalancha de datos de significación incierta, por lo que resultará imprescindible la descripción depurada de las características clínicas y radiológicas de los pacientes.
El consejo genético debe realizarse por especialistas preparados y aportar información al paciente y/o a la familia sobre los riesgos que presenta la enfermedad y posibles tratamientos, así como el riesgo de transmisión a su descendencia, entre otros. En cuanto al consejo genético en cavernomatosis, es importante tener en cuenta los siguientes puntos: a)identificación de la mutación; b)establecimiento de si es una forma familiar o una forma esporádica de aparición en línea germinal; c)penetrancia clínica y estudio en colaboración con el clínico, quien deberá estudiar si presenta una sola malformación o una cavernomatosis múltiple; d)ofrecimiento de estudio molecular a familiares de primer grado, teniendo en cuenta que es el familiar el que debe insinuar la idea, después que haya habido una explicación del riesgo propio y de otros familiares; e)iniciar siempre el estudio en los padres del posible probando. Esto es particularmente importante en niños de riesgo, en los que el consejo genético se debe hacer con mayor cautela; f)en general es una enfermedad muy peculiar a la hora del consejo genético debido a la posibilidad de un tratamiento sintomático e incluso de extirpación quirúrgica de la lesión, y g)no ofrecer consejo genético mediante posible diagnóstico molecular en embarazadas cuando no se ha identificado la mutación.
ConclusionesEl diagnóstico molecular de la cavernomatosis cerebral debe incluir al menos la detección del número de copias y la secuenciación de los genes CCM. En el caso de no encontrar la mutación responsable de la enfermedad, podrían llevarse a cabo otras alternativas. La detección de mutaciones no es completa, por lo que el estudio molecular no puede ser definitivo y empleado para descartar la enfermedad. Finalmente, ofrecer un adecuado consejo genético es importante para proporcionar información y apoyo a los pacientes y/o familias que padecen la enfermedad.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

