



La enfermedad de Danon es un raro trastorno de herencia dominante ligada al cromosoma X, caracterizado por la tríada de discapacidad intelectual miopatía y miocardiopatía hipertrófica. Puede asociar variable retinopatía u otra afectación ocular. Inicialmente fue considerada como una variante de la glucogenosis tipo II con actividad normal de maltasa ácida1. Posteriormente se observó que no siempre se acumulaba glucógeno y que había una deficiencia de una proteína estructural de los lisosomas (LAMP-2) cuyo papel no se conoce bien2. Avances más recientes pusieron de manifiesto que esta enfermedad es una forma de miopatía autofágica vacuolar. Las vacuolas son autolisosomas rodeados de proteínas sarcolémicas, lámina basal y actividad de acetil colinesterasa3. Hasta la fecha se han descrito más de 30 mutaciones diferentes del gen de la proteína LAMP-2 y con diferentes mecanismos de acción4.
Nosotros hemos estudiado una familia con madre e hijo afectados por esta enfermedad y encontramos una mutación nueva de dicho gen.
Niño con historial de deterioro psicomotor tras un desarrollo normal desde los 7 años, así como torpeza de extremidades y síntomas de intolerancia al ejercicio. En la exploración general había epicantus, paladar ojival y pabellones auriculares alados. Sufría síntomas de hiperactividad y bulimia que mejoraron con metilfenidato. En la analítica había elevación constante de la CPK (entre 400 y 1.000U/l) y de las transaminasas GOT y GPT (por encima de 200U/l). El EMG fue normal. El cariotipo y el estudio genético del cromosoma X frágil no revelaron alteraciones. El estudio por MLPA de mutaciones de la distrofina no mostró deleciones ni duplicaciones. El test de la gota seca para la enfermedad de Pompe fue negativo. Tras estudios cardiológicos normales, desarrolló un QRS estrecho con amplitud aumentada de las ondas R en precordiales en el ECG, presentó episodios de taquicardia y el ecocardiograma evidenció una miocardiopatía hipertrófica con función ventricular conservada.
Su madre padeció una miocardiopatía dilatada posparto y necesitó trasplante cardíaco, pero en su momento no se conocía la causa que la originó. El paciente tiene una hermana asintomática.
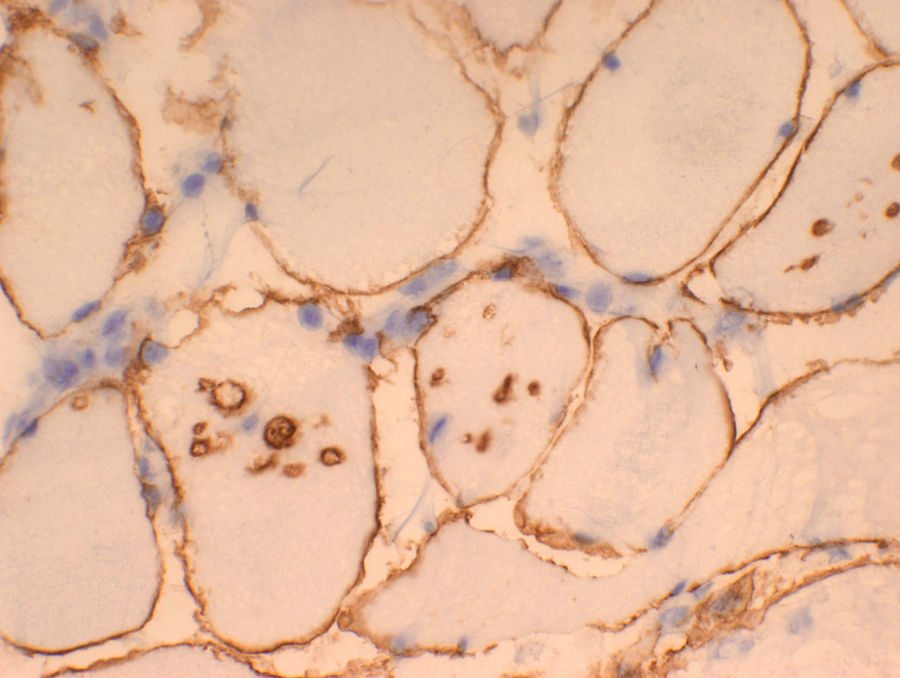
Se realizó biopsia muscular a los 11 años, y en la muestra se objetivó la presencia de vacuolas basófilas rodeadas de proteínas sarcolémicas como distrofina, utrofina, emerina y sarcoglicanos (fig. 1). No se encontraron acúmulos de glucógeno. La inmunohistoquímica para la proteína LAMP-2 resultó negativa, pero con algún artefacto de descongelación. Seguidamente se procedió al estudio genético molecular con secuenciación automática y electroforesis capilar (Hospital Meixoeiro de Vigo) encontrándose una mutación en el en el exón 8 del gen LAMP-2, A314GfsaX2, no descrita previamente. En la madre se encontró la misma mutación, pero en la hermana el estudio genético fue normal. Actualmente tiene 15 años, su situación clínica está estable y sigue controles con asiduidad por los servicio de cardiología y neurología.

La enfermedad de Danon tiene una prevalencia menor de un caso por millón. El diagnóstico se lleva a cabo a raíz de los síntomas neurológicos o cardiológicos. En la serie más larga publicada hasta ahora con 82 casos se encontró retraso mental en el 100%, miocardiopatía en el 88% y debilidad muscular en el 80%. Los síntomas son menos severos en las mujeres que en los varones y comienzan más tarde. Combinando estos 82 casos con otros 63 publicados previamente se observó que las edades del primer síntoma, trasplante cardíaco y muerte fueron 12, 18 y 19 años en los varones, y 28, 33 y 34 en las mujeres, respectivamente4.
La sospecha clínica de esta enfermedad es importante porque los problemas cardiológicos se pueden tratar5. En un estudio de 50 pacientes con miocardiopatía hipertrófica y estudio genético negativo para mutaciones en 9 genes sarcoméricos se encontraron 2 casos con enfermedad de Danon, lo que supone un 1% de los 197 pacientes iniciales que pasaron un cribado de miocardiopatía hipertrófica6.
La existencia de hiperCKemia con discapacidad intelectual se da en las distrofinopatías, especialmente la enfermedad de Duchenne, y en la enfermedad de Danon, y puede haber discreta elevación de CK en la distrofia miotónica congénita. Sin embargo deben contemplarse otras causas, pues dada la elevada prevalencia de la discapacidad intelectual, puede darse su asociación casual con cualquier otra miopatía.
Destacamos la importancia del estudio de las hiperCKemias persistentes significativas, que en ausencia de diagnóstico debe incluir la biopsia muscular. Destacamos así mismo la necesidad del control cardiológico en diversas miopatías que pueden asociar miocardiopatía y la vigilancia de miopatías en familias con cardiomiopatías sin diagnóstico etiológico establecido.

