






La esclerosis lateral amiotrófica (ELA) es una enfermedad con muy mal pronóstico, con una mortalidad del 50% a los 18 meses tras el diagnóstico. Las unidades multidisciplinares pretenden mejorar la calidad de vida y la supervivencia de los enfermos de ELA. El objetivo de nuestro estudio es evaluar cada 3 meses la evolución de pacientes atendidos en la unidad de ELA desde el momento del diagnóstico y durante 24 meses.
Material y métodosSe realizó un estudio observacional prospectivo de pacientes atendidos en la unidad de ELA siguiendo una vía clínica desde el momento del diagnóstico y con revisiones trimestrales desde 2006 a 2010. La edad de inicio, el deterioro de la situación funcional (escala ALSFRS-r), el deterioro de la función respiratoria y la aparición de disfagia y de signos de depresión y/o de deterioro cognitivo fueron evaluados en relación con la localización inicial de los síntomas (bulbar [B], miembros superiores [MMSS], miembros inferiores [MMII]).
Resultados42 pacientes (30V y 12M) fueron evaluados (edad media de inicio±desviación estándar de 57,97±14,56 años). Se encontró una distribución igual por localización de inicio de los síntomas (B 14 pacientes, MMSS 14, MMII 14). El deterioro funcional (B –26,89 pts.; MMSS –22,48 pts.; MMII –22,66 pts.), la necesidad de uso de BIPAP (B 64,28%; MMSS 35,71%, MMII 50%), la presencia de disfagia (B 85,71; MMSS 42.85; MMII 71.42%), de signos de depresión (B 78,57%, MMSS 35,71%; MMII 64,28%) y de deterioro cognitivo (B 42,85%; MMSS 21,42; MMII 35,71%) fue mayor a los 24 meses de evolución en los pacientes de inicio bulbar. No hubo diferencias en los datos de mortalidad (global 23,80%).
ConclusionesEl tratamiento en unidades multidisciplinares no varía la evolución neurológica de la enfermedad pero favorece la aplicación de cuidados multidisciplinares e incrementa la supervivencia de los enfermos de ELA independientemente de su forma de inicio.
Amyotrophic lateral sclerosis (ALS) is a disease with very poor prognosis, and a mortality of 50% at 18 months after diagnosis. Multidisciplinary units attempt to improve the quality of life and survival of patients with ALS. The aim of this study is to evaluate every 3 months, over a 24-month period, the outcome of patients treated at the ALS unit since the time of diagnosis.
Material and methodsWe performed a prospective observational study of patients treated in the ALS unit following a clinical pathway since the time of diagnosis with quarterly reviews from 2006 to 2010. The age of onset, functional impairment (ALSFRS-r), impairment of respiratory function, dysphagia and signs of depression and/or cognitive impairment were evaluated in relation to the initial location symptoms (bulbar [B], upper limbs [UL], lower limbs [LL]).
ResultsA total of 42 patients (30 males and 12 females) were evaluated (mean age at onset of 57.97years old, SD 14.56). There was an even distribution by location of onset of symptoms (B 14 patients, UL 14, LL 14.) Functional impairment (B –26,89 points, UL –22,48 points, LL –22,66 points), the need for use of BIPAP (B 64.28%; UL 35.71%; LL 50%), the presence of dysphagia (B 85.71; UL 42.85; LL 71.42%), signs of depression (B 78.57%; UL 35.71%; LL 64.28%) and cognitive impairment (B 42.85%; UL 21.42; LL 35.71%) was higher at 24 months of progression in patients with bulbar onset. There was no difference in mortality data (23.80% overall).
ConclusionsThe treatment in multidisciplinary units does not change the neurological progression of the disease, but increases the survival of ALS patients regardless of their initial onset, emphasising the use of multidisciplinary care.
La esclerosis lateral amiotrófica (ELA) es una enfermedad neurodegenerativa caracterizada por presentar signos y síntomas de degeneración primaria de las motoneuronas superior e inferior. En el curso de la enfermedad se produce una debilidad y una atrofia progresivas de la musculatura de inervación bulbar, torácica, abdominal y de las extremidades.
El riesgo de desarrollar una ELA a lo largo de la vida es de 1:1000. La incidencia de la enfermedad se cifra entre 1,5 y 2,7 por cada 100.000 habitantes/año en Europa y Norte América con un leve predominio en varones (1.5:1) y una edad media de inicio sintomático de 64 años. El fallecimiento suele deberse a un fallo respiratorio que se produce entre los 3 y los 5 años desde el inicio de los síntomas; según series históricas el 50% fallece a los 18 meses del diagnóstico1–4.
Actualmente, la ELA carece de un tratamiento curativo contando solamente con el riluzol como terapia específica para aumentar levemente la supervivencia. Su manejo se basa esencialmente en el tratamiento paliativo y el control de los síntomas5–7, incluido el uso de fármacos y la implantación de gastrostomías percutáneas y de sistemas de ventilación invasiva o no invasiva.
En los últimos años las unidades multidisciplinares para el tratamiento de estos enfermos han emergido en todo el mundo7,8. La acumulación de un gran número de pacientes lidera la agrupación de recursos y de expertos clínicos que facilitan el tratamiento de esta enfermedad. Aunque las unidades multidisciplinares mejoran la calidad de vida y prolongan la supervivencia de otras enfermedades neurodegenerativas, su efecto en la ELA aún no está claro7–11.
Una red de unidades multidisciplinares de ELA especializada en el diagnóstico, manejo y cuidados paliativos se puso en marcha en Madrid (España) en 2006. El cuidado de los enfermos se protocolizó mediante el desarrollo de una vía clínica que incluía aspectos de diagnóstico, tratamiento y cuidados, tanto desde el punto de vista médico como de la atención social12.
El objetivo del presente estudio es analizar los datos de los pacientes de ELA tratados en una de las unidades de la red bajo el protocolo de la vía clínica desarrollada cada 3 meses durante los 2 primeros años tras el diagnóstico.
Material y métodosRealizamos un estudio observacional descriptivo de pacientes atendidos en la unidad multidisciplinar de ELA desde marzo de 2006 hasta enero de 2010.
Se seleccionó exclusivamente para el estudio a aquellos enfermos diagnosticados de ELA en categoría de definitivo o probable según los criterios revisados de El Escorial13, que acudieron de forma regular, al menos cada 3-4 meses, a revisión en la unidad multidisciplinar. Se excluyó a los pacientes que no completaron el seguimiento al menos un año tras el diagnóstico. El periodo máximo de seguimiento del estudio fue de 2 años.
Todos los pacientes seleccionados fueron tratados con riluzol durante el periodo de seguimiento, aunque éste no fue un factor de inclusión/exclusión en estudio. En la unidad multidisciplinar, siguiendo la vía clínica12 establecida, recibieron atención neurológica, psicológica, paliativa y social; se realizan estudios respiratorios y nutricionales adaptando gastrostomías o sistemas de ventilación no invasiva y se realiza tratamiento de rehabilitación motriz, respiratoria y foniátrica según las necesidades del enfermo desde el momento del diagnóstico.
Se analizaron los datos de edad y la localización inicial de los síntomas, el deterioro funcional trimestral (mediante la escala ALSFRS-r14), el deterioro de la función respiratoria (FVC<70%), la aparición de disfagia (escala Karnell15 mayor de 2) y de signos de depresión (más de veintiún puntos de la escala BDI16) y/o deterioro cognitivo (MEC<24/30) durante los 2 primeros años tras el diagnóstico.
La instauración de sistemas de ventilación no invasiva y de gastrostomías no se determinó por ningún test o prueba única, sino por la conjunción de distintas variables que en cada enfermo pudieron ser distintas (caída FVC, índice de desaturación en pulsioximetría nocturna, valores del PIM y PEM para los sistemas de ventilación) (pérdida de peso rápida, disfagia intensa, deshidratación para las gastrostomías).
Estos datos fueron evaluados en relación a la localización inicial de los síntomas de la siguiente manera: bulbar (B), miembros superiores (MMSS) o miembros inferiores (MMII).
El estudio estadístico fue realizado mediante el programa informático estadístico, PASW Statistics 18 usando el test de la χ2 para las diferencias entre las variables categóricas y el test de McNemar para las variables pareadas, y generando un modelo de análisis de medias de mínimos cuadrados. Se consideran significativos valores de p < 0,05.
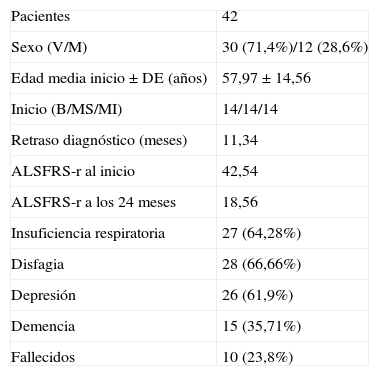
ResultadosCuarenta y dos enfermos diagnosticados de ELA en categoría de definitivo o probable fueron seleccionados siguiendo los criterios expuestos previamente de entre los 97 atendidos en la unidad de ELA entre marzo de 2006 y enero de 2010. De ellos, 30 eran varones y 12 mujeres, la edad media de inicio de los síntomas±desviación estándar fue de 57,97±4,56 años y el retraso hasta el diagnóstico se pudo cifrar en 11,34 meses (tabla 1).
Distribución de la muestra
| Pacientes | 42 |
| Sexo (V/M) | 30 (71,4%)/12 (28,6%) |
| Edad media inicio±DE (años) | 57,97±14,56 |
| Inicio (B/MS/MI) | 14/14/14 |
| Retraso diagnóstico (meses) | 11,34 |
| ALSFRS-r al inicio | 42,54 |
| ALSFRS-r a los 24 meses | 18,56 |
| Insuficiencia respiratoria | 27 (64,28%) |
| Disfagia | 28 (66,66%) |
| Depresión | 26 (61,9%) |
| Demencia | 15 (35,71%) |
| Fallecidos | 10 (23,8%) |
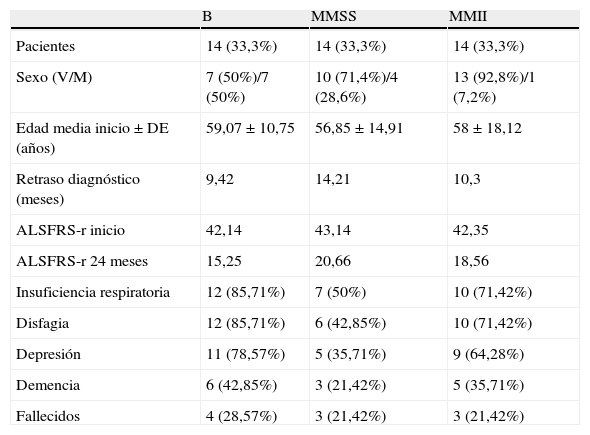
Se encontró una distribución igual en los 3 grupos creados según la localización inicial de los síntomas por localización de inicio de los síntomas (14 pacientes en cada grupo). Los pacientes del grupo B eran 7 varones (50%) y 7 mujeres (50%), con una edad media de inicio de los síntomas de 59,07 años (V 55,57 vs M 62,57). Los del grupo MMSS era de 10 varones (71,42%) y 4 mujeres (28,57%), con una edad media al inicio de 56,85 años (V 57,1 vs M 56,25). Por último, los del grupo MMII eran 13 varones (92,85%) y 1 mujer (7,14%), y la edad de inicio fue de 58,74 años (V 57,92 vs M 59). Las diferencias de edad entre los grupos no fueron estadísticamente significativas (p>0,05). El retraso diagnóstico fue menor en los pacientes de inicio bulbar, pero sin diferencias significativas (tabla 2).
Distribución de muestra por grupos
| B | MMSS | MMII | |
| Pacientes | 14 (33,3%) | 14 (33,3%) | 14 (33,3%) |
| Sexo (V/M) | 7 (50%)/7 (50%) | 10 (71,4%)/4 (28,6%) | 13 (92,8%)/1 (7,2%) |
| Edad media inicio±DE (años) | 59,07±10,75 | 56,85±14,91 | 58±18,12 |
| Retraso diagnóstico (meses) | 9,42 | 14,21 | 10,3 |
| ALSFRS-r inicio | 42,14 | 43,14 | 42,35 |
| ALSFRS-r 24 meses | 15,25 | 20,66 | 18,56 |
| Insuficiencia respiratoria | 12 (85,71%) | 7 (50%) | 10 (71,42%) |
| Disfagia | 12 (85,71%) | 6 (42,85%) | 10 (71,42%) |
| Depresión | 11 (78,57%) | 5 (35,71%) | 9 (64,28%) |
| Demencia | 6 (42,85%) | 3 (21,42%) | 5 (35,71%) |
| Fallecidos | 4 (28,57%) | 3 (21,42%) | 3 (21,42%) |
En el seguimiento trimestral de los pacientes se pudo objetivar cómo el declive funcional es significativo estadísticamente a partir de los 6 meses de seguimiento (p=0,0297). La situación funcional (escala ALSFRS-r) de los enfermos inicial es de 42,54 puntos y se deteriora hasta los 18,07 a los 24 meses (–24,47 puntos) (fig. 1).

Como se puede observar en la figura 2, la situación funcional inicial de los pacientes del grupo B es de 42,14 puntos ALSFRS-r de media frente a 43,14 y 42,35 puntos de los de los grupos MMSS y MMII. A los 24 meses, la situación funcional también es peor que en el resto de los grupos (B 15,25 vs MMSS 20,66 vs MMII 19,69) y, por tanto, el decremento promedio en estos 24 meses también es más acentuado (B –26,89 vs MMSS –22,48 vs MMII –22,66) (p>0,5). En el deterioro es significativo en los distintos grupos a partir de los 12 meses (grupo B p=0,0059; MMSS p=0,0197; MMII p=0,0003). Las diferencias entre los grupos a estudio no son estadísticamente significativas (p=0,9971).

Los síntomas de insuficiencia respiratoria afectaron al 69,04% de los enfermos, siendo indicada la instauración de una ventilación mecánica no invasiva en el 54,76% del total de pacientes, el 79,3% de aquellos cuya CVF fue inferior al 70%.
Por grupos, 12 (85,71%) de los enfermos del grupo B sufrieron un decremento significativo de la función respiratoria, frente a 7 (50%) (p=0,04) de los pacientes del grupo MMSS y 10 (71,42%) (p=0,43) del grupo MMII a lo largo de los 2 años de seguimiento (fig. 3). Además, se indicó que debían iniciar tratamiento con un dispositivo de ventilación mecánica no invasiva, la usaran finalmente o no, a 9 (64,28%) de los enfermos del grupo B, 5 (35,71%) del MMSS y 7 (50%) del MMII. Estos datos no son significativos de forma global (p=0,309).

Presentaron disfagia 28 pacientes (66,6%), 12 de los enfermos del grupo B (85,71%) frente al 42,85% (6 enfermos) del grupo MMSS y al 71,42% (10 pacientes) del grupo MMII (fig. 3) (p=0,355). Se implantó una gastrostomía al 46,42% de los enfermos con disfagia (30,95% del global).
En cuanto a los síntomas de depresión o de alteración cognitiva, también fueron más importantes en el grupo B. El 78,57% (11) de los enfermos del grupo B presentaron síntomas depresivos de forma reactiva desde el inicio de la enfermedad. Cinco de estos 11 enfermos también tenían signos de deterioro cognitivo y también hubo otro caso sin síntomas depresivos asociados. Los 6 pacientes con signos de deterioro cognitivo representan el 42,85%.
En el grupo MMSS 5 pacientes (35,71%) (p=0,250) tuvo síntomas depresivos, asociándose en 3 signos deterioro cognitivo (21,42%), en el grupo MMII había 9 (64,28%) (p=0,50) con síntomas depresivos y 5 (35,71%) de ellos con signos de deterioro cognitivo (fig. 3) (p=0.87).
En los datos de mortalidad no hubo grandes diferencias, 4 enfermos (28,57%) fallecieron en el grupo B, 3 (21,42%) en el MMSS y 3 (21.42%) en el MMII a lo largo de los 2 años de estudio (p>0,05), lo que significa una mortalidad global del 23,80%.
DiscusiónLos datos anteriores a la creación de la unidad de ELA se encuentran muy incompletos y sesgados. Los pacientes en muchas ocasiones no eran seguidos tras el diagnóstico y no existen datos sobre la evolución de su situación funcional neurológica o neumológica ni valoración neuropsicológica. Recurriendo a los registros de la farmacia de nuestro hospital, pudimos en su momento analizar los informes de aquellos enfermos que habían estaban tomando riluzol en los años anteriores al desarrollo de la unidad17. En esos datos podemos ver cómo el retraso diagnóstico ha disminuido de 27,61 a 11,34 meses desde el inicio de los síntomas y que la implantación de sistemas de ventilación (11,5% preunidad) y de gastrostomías (8,6% preunidad) y el porcentaje de enfermos atendidos por síntomas de depresión (10,6% preunidad) era mucho menor que los datos que obtenemos actualmente. No existen datos fiables de mortalidad en esa etapa preunidad.
Aunque no era el objeto del estudio, lo que más llama la atención del registro de pacientes de la unidad de ELA son los datos de mortalidad. La cifra de fallecidos a los 24 meses de seguimiento es claramente inferior que las que aparecen en los artículos de revisiones del curso clínico de la enfermedad donde se cifraba la mortalidad en un 50% a los 18 meses1–4 y que los datos objetivados en distintos trabajos diseñados para evaluar si las unidades multidisciplinares eran útiles para mejorar la supervivencia de los pacientes9,11,18.
Así, en el estudio realizado en las unidades multidisciplinares de Irlanda18 se concluyó que los pacientes de ELA atendidos en unidades multidisciplinares frente a aquellos que eran tratados en servicios de neurología general fallecían más tardíamente y sobre todo se beneficiaban de los cuidados multidisciplinares los enfermos cuyos síntomas se habían iniciado a nivel bulbar. Pese a ello, la mortalidad a los 2 años estaba en torno al 50% de forma global y del 57% en los casos bulbares.
En el estudio realizado por Chio9 para valorar la repercusión en el pronóstico de la enfermedad de la atención y las visitas continuadas a un hospital terciario, la supervivencia media de los pacientes atendidos en un centro terciario de ELA es de 1.080 días, pero no hay datos de porcentaje de mortalidad a los 2 años.
Incluso en el estudio que se realizó en unidades multidisciplinares del sur de Italia11 los datos eran peores, con una mortalidad en los primeros 12 meses del 24% en pacientes. En este trabajo destacaba que más del 30% de los enfermos no estaban en tratamiento con riluzol.
Por tanto, nuestros datos con una mortalidad a los 24 meses de tan sólo el 23% de los enfermos, 29% en el caso de los de inicio bulbar, son claramente mejores. Esto pudiera explicarse por varios factores.
Nuestros enfermos son más jóvenes que los de los otros estudios9,11,18 (58 años frente a 60, 61 y 64 años, respectivamente). Distintos trabajos19–21 han evaluado la historia natural de la ELA en pacientes jóvenes, concluyendo que en esos casos tiene una mayor supervivencia. Sin embargo, en nuestro trabajo los enfermos no entran en la categoría de «joven» aplicada en estos estudios en los donde se recogían pacientes de menos de 2519 o de 4019–21 años y la diferencia de las medias de edad respecto a los estudios de otras unidades multidisciplinares9,11,15 es muy escasa. Por otro lado, en estos trabajos previos los pacientes además de más jóvenes tenían un predominio de los síntomas de motoneurona superior o un importante intervalo desde el inicio de los síntomas hasta el diagnóstico, cuestiones que no se cumplen en nuestro estudio.
Quizá el factor fundamental que deberíamos considerar es el estricto seguimiento de los enfermos protocolizado que desarrollamos en nuestro medio12. La red de unidades irlandesa18 también tiene un seguimiento intenso de los pacientes, pero es cada 6 meses y, en ocasiones, por contacto telefónico, por lo que no es posible asegurar los cuidados de algunos de los aspectos de la enfermedad. Por otro lado es el único que tiene una cuidada red de asistencia social-paliativa como la nuestra. Los estudios italianos9,11 hablan de la atención en unidades pero sin contar con un protocolo de tiempos estándar en el seguimiento.
Estos datos estarían en consonancia con las recomendaciones de la AAN y de la EFNS22,23 acerca de que el seguimiento protocolizado podría optimizar la atención al paciente y mejorar su supervivencia. Así la instauración de sistemas de ventilación en pacientes con insuficiencia respiratoria y de gastrostomías fue claramente superior a la recogida en los primeros registros24 tras la publicación de las guías clínicas de la AAN y podría explicar la mayor supervivencia de nuestros enfermos.
A nivel funcional neurológico no se podría hablar de diferencias entre los grupos aunque existe una leve tendencia a un mayor decremento en la puntuación de la escala ALSFRS-r en el grupo B. Las curvas de deterioro no son muy diferentes de las observadas por Gordon recientemente en pacientes de ELA no seguidos en unidades multidisciplinares25.
Respecto al resto, los resultados de la afectación multidisciplinar observamos una tendencia a un peor pronóstico respiratorio y la mayor necesidad de cuidados de los pacientes con clínica de inicio bulbar respecto a los enfermos de inicio espinal aunque el pequeño tamaño de la muestra no permite obtener diferencias significativas estadísticamente. Llama la atención que los resultados obtenidos en los pacientes con afectación inicial de los MMII sea peor que aquellos que comenzaron en los MMSS, aunque las diferencias son pequeñas.
El peor pronóstico de los pacientes de afectación bulbar se ha evidenciado y ha sido tratado en varios trabajos26–28, donde se ha llegado a estimar la existencia de factores de confusión, como podría ser la edad de inicio de los síntomas, por ser habitualmente más mayores estos pacientes. En nuestro caso no existía una clara diferencia de edad entre los grupos y, por tanto, sí nos parece muy valorable la tendencia a una peor evolución de los enfermos con clínica bulbar respecto a los de predominio espinal. Sin embargo, no podríamos decir que exista un incremento de mortalidad en esos pacientes respecto a los espinales durante los dos primeros años.
Pensamos que los altos niveles de pacientes con síntomas de depresión y de deterioro cognitivo se deben a que los pacientes son seguidos en una unidad multidisciplinar12 y el seguimiento protocolizado por los distintos especialistas podría facilitar el hallazgo subclínico de estos datos de forma más frecuente que en otros trabajos29,30.
En conclusión, podemos afirmar que el tratamiento en unidades multidisciplinares no parece variar la evolución neurológica de la enfermedad pero favorece la aplicación de cuidados respiratorios y nutricionales, así como la detección y atención de los síntomas de depresión y deterioro cognitivo. Gracias a todo esto, la supervivencia de los pacientes con ELA tratados en una unidad multidisciplinar es mayor, independientemente de su forma de inicio.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

