




Existen algunas especies de primates no humanos con algunas de las características definitorias de la enfermedad de Alzheimer (EA) del hombre, tanto en el aspecto neuropatológico como en el cognoscitivo-comportamental, y que son de importancia capital para entender y/o tratar esta enfermedad.
DesarrolloEn esta segunda parte del estudio se analizan estas características durante el proceso de envejecimiento en los modelos de EA más importantes de primates no humanos no experimentales (lémur ratón —Microcebus murinus—, cercopiteco verde —Chlorocebus aethiops— y los macacos Rhesus y de cola en tirabuzón —Macaca mulatta y M. arctioides—) y experimentales (modelos lesionales, neurotóxicos, farmacológicos, inmunológicos, etc.). En todos estos modelos se puede presentar neuropatología amiloidea cerebral senil, aunque con diferente grado de incidencia (100% en cercopitecos; < 30% en macacos). Las diferencias entre senilidad normal y patológica (Alzheimer) en estas especies son difíciles de establecer por la falta de estudios cognoscitivo-comportamentales en muchos grupos analizados, así como por la controversia existente en los resultados de estos estudios cuando se llevaron a cabo. Sin embargo, en algunos macacos se ha comprobado la correlación entre un alto grado de deterioro funcional cerebral y una gran cantidad de alteraciones neuropatológicas (posible «Alzheimer»).
ConclusionesEn los macacos, se puede considerar la existencia de un posible «continuum» entre proceso de envejecimiento «normal», «normal con no profundas alteraciones neuropatológicas y cognoscitivo-comportamentales», y «envejecimiento patológico» o «envejecimiento tipo Alzheimer». En otros casos, como el de los cercopitecos verdes, las alteraciones neuropatológicas son constantes y bastante marcadas, pero sus repercusiones cognoscitivo-comportamentales no parecen muy importantes. Ello hace suponer la posible existencia en la involución senil fisiológica de una fase estable sin demencia aun cuando existan alteraciones neuropatológicas.
In the ageing process there are some species of non-human primates which can show some of the defining characteristics of the Alzheimer's disease (AD) of man, both in neuropathological changes and cognitive-behavioural symptoms. The study of these species is of prime importance to understand AD and develop therapies to combat this neurodegenerative disease.
DevelopmentIn this second part of the study, these AD features are discussed in the most important non-experimental AD models (Mouse Lemur —Microcebus murinus, Caribbean vervet —Chlorocebus aethiops, and the Rhesus and stump-tailed macaque —Macaca mulatta and M. arctoides) and experimental models (lesional, neurotoxic, pharmacological, immunological, etc.) non-human primates. In all these models cerebral amyloid neuropathology can occur in senility, although with different levels of incidence (100% in vervets;<30% in macaques). The differences between normal and pathological (Alzheimer's) senility in these species are difficult to establish due to the lack of cognitive-behavioural studies in the many groups analysed, as well as the controversy in the results of these studies when they were carried out. However, in some macaques, a correlation between a high degree of functional brain impairment and a large number of neuropathological changes (“possible AD”) has been found.
ConclusionsIn some non-human primates, such as the macaque, the existence of a possible continuum between “normal” ageing process, “normal” ageing with no deep neuropathological and cognitive-behavioural changes, and “pathological ageing” (or “Alzheimer type ageing”), may be considered. In other cases, such as the Caribbean vervet, neuropathological changes are constant and quite marked, but its impact on cognition and behaviour does not seem to be very important. This does assume the possible existence in the human senile physiological regression of a stable phase without dementia even if neuropathological changes appeared.
Existen una serie de especies de primates no humanos que por diversos motivos (corta expectativa de vida, acumulación de amiloide, uso habitual en el laboratorio, etc.) han sido estudiadas con mayor detenimiento y han sido propuestas como posibles «modelos animales» para el estudio del envejecimiento cerebral y la enfermedad de Alzheimer (EA)1. Presentan características distintas, con similitudes y diferencias frente al humano con EA, que se precisan conocer antes de extrapolar al humano las conclusiones de los estudios llevados a cabo en ellas. Las especies más estudiadas en sus aspectos neuropatológicos corresponden las familias Cherirogaleidae (lémures, pro-simios) y Cercopithecoideae (simios cercopitecos) (véase parte I1). Los grandes simios evolutivamente más próximos al hombre (especialmente gorila, en libertad, y chimpancé, en cautividad) han sido objeto, hasta hace pocos años, de importantes investigaciones sobre el comportamiento y las funciones cerebrales en un sentido amplio del tema, pero estos estudios están bastante alejados de la problemática que nos ocupa, la involución senil morfofuncional normal o patológica del cerebro.
Lémures (prosimios)La especie más estudiada como modelo animal de EA es Microcebus murinus (lémur ratón)2-8. Es un mamífero parecido a un roedor, originario de Madagascar, pero que se ha criado en múltiples laboratorios de todo el mundo. Tiene la ventaja de ser pequeño (entre 12 y 13cm de longitud corporal, con una cola igualmente larga y entre 50 y 100g de peso) y tener una esperanza de vida corta, ya que los animales con más de 5 años se consideran ancianos. Sin embargo, en algunos grupos en cautividad se ha señalado una longevidad de hasta 14 años. La mayoría de los lémures presenta un envejecimiento cerebral progresivo, lento y sin gran involución, mientras que el 20% desarrolla neurodegeneración temprana. En estos primates, la neurodegeneración se caracteriza por atrofia cerebral masiva en corteza, hipocampo, ganglios basales, troncoencéfalo y cerebelo, asociada con un aumento considerable del tamaño de los ventrículos, abundantes placas amiloideas, acumulaciones de proteína tau y pérdida de neuronas colinérgicas. Además de las placas, presentan otros tipos de depósitos amiloideos y de filamentos argirófilos neuronales, tal como ocurre en muchos pacientes con EA. Los animales que sufren esta neurodegeneración pierden capacidades cognoscitivo-comportamentales (realización de tareas y relaciones sociales) que muestran ciertas similitudes con los cambios comportamentales desarrollados en EA humana. Los estudios realizados sobre la composición, maduración y distribución de las placas amiloides en Microcebus murinus también han demostrado ciertas similitudes con el humano. En Microcebus, el β-amiloide se deposita de forma que puede asemejarse a lo que se observa en humanos. Existen tres tipos principales de acumulaciones: placas difusas de unas 100 micras de diámetro, placas densas de núcleo compacto de amiloide, que son además tioflavina positivas, y formaciones en forma de cinta alrededor de los vasos sanguíneos; los dos primeros tipos de placas también se encuentran en enfermos de EA. Al igual que en el hombre, los depósitos amiloideos de Microcebus tienen dos componentes principales, Abeta42 y Abeta40, demostrable mediante técnicas inmunocitoquímicas, y se observó que los depósitos sobre los vasos corticales eran inmunopositivos para ambos componentes, que las acumulaciones difusas eran altamente positivas para Abeta42 y que algunas placas solo eran positivas para Abeta42. Esto sugiere que Abeta42, de la misma manera que se observa en humanos, está asociada a etapas tempranas de la maduración de las placas9. La relación Abeta40/Abeta42 es mayor en este primate que en humanos, es decir, que existe una pequeña diferencia en cuanto a los mecanismos de producción de amiloide9. La distribución de placas en Microcebus, de mayor a menor densidad, es la siguiente: parte superior del lóbulo temporal, amígdala, área prefrontal, lóbulo parietal y lóbulo occipital. Por tanto, el patrón de afectación amiloidea es, en parte, similar al del humano3,4. Se han realizado estudios de secuenciación del gen que codifica APP que han revelado que, aun existiendo ciertas diferencias en algunos nucleótidos, la proteína y el péptido Abeta resultante de su catabolización son completamente homólogos a los humanos3. APP se localiza en los cuerpos celulares y dendritas proximales de neuronas, en astrocitos y oligodendrocitos y en los depósitos amiloideos del parénquima cortical y de las paredes de los vasos sanguíneos de manera parecida a lo que ocurre en el hombre. Además, se observó una buena correlación entre niveles de APP y densidad de placas amiloides, aumentando ambos con la edad del animal3,4.
Otra de las características que definen la EA en humanos, la acumulación de proteína tau, se ha encontrado también en Microcebus murinus, donde aumenta de forma constante con la edad. El área más afectada es la neocorteza, incluso en animales jóvenes, mientras que el subículo y la corteza entorrinal sólo se ven afectados en animales mayores de 8 años (ya considerados muy ancianos), todo ello muy diferente de lo observado en humanos. Aunque todos los animales que presentaban beta-amiloidosis difusa también tenían acumulaciones de tau en la neocorteza, no se encontró correlación entre las densidades de estas lesiones en cada área con el resto de las áreas estudiadas3,4.
Cercopitecos (simios del viejo mundo)Los géneros Chlorocebus y Macaca acaparan la atención de la investigación.
Chlorocebus aethiops (mono verde o cercopiteco verde), de origen africano y presente actualmente en varios países de Asia y América, y en concreto sus colonias residentes en la isla de St. Kitts en el Caribe, ha sido propuesto recientemente como un modelo para el estudio de la patología amiloidea en el Alzheimer10-12. Los machos adultos pesan de 5 a 7kg y viven de 20 a 30 años cuando están en cautividad (véase sección 4.1, parte I). Estudios realizados en ejemplares de 15, 22 y 30 años muestran que a los 15 años comienzan a aparecer depósitos amiloideos (predominantemente de Abeta40) sobre los vasos sanguíneos, aunque no existan placas en el parénquima cerebral. En todos los ejemplares de 22 y 30 años (en estos últimos muy abundantemente), se encuentran placas en las cortezas frontal, temporal y occipital, siendo, inmunohistoquímicamente, Abeta42 mucho más abundante que Abeta40, tal como se aprecia en cerebros de humanos que han padecido EA. El amiloide fibrilar, demostrado mediante tinción con tioflavina S, se detecta sobre los vasos a las tres edades, más a nivel occipital, lo que no concuerda con el estudio en humanos, pero sólo en una pequeña proporción de placas a los 22 y 30 años (también a diferencia con el humano). A los 15 años de edad no parecen existir alteraciones neuríticas ni tampoco gliales y solo en los monos de 22 años aparecieron algunas placas de apariencia neurítica. Sin embargo, todos los monos de 30 años mostraron una amplia variedad de alteraciones neuropatológicas típicamente asociadas a las placas de la EA humana (astrocitos reactivos, microglía activada, neuritas distróficas). La gran cantidad de placas neuríticas a esta edad, cuya densidad era parecida a la observada en los humanos que padecieron EA, contrasta con la escasez de estas estructuras en la mayoría de los primates no humanos antes estudiados10-12. Sin embargo, aunque estas placas eran inmunopositivas para sustancias amiloideas diversas, incluido APP, solo un pequeño porcentaje lo eran para proteína tau fosforilada, característica humana de neuritas distróficas. Lo mismo ocurre con los ovillos neurofibrilares intraneuronales, que se mostraron muy diferentes, morfológica e histoquímicamente, de los humanos. Esto último plantea importantes problemas teórico-prácticos, que se discutirán más adelante.
Dentro del género Macaca están los primates no humanos más empleados en la investigación científica y que también acaparan el mayor número de publicaciones sobre el tema de la senilidad normal y patológica en el cerebro1. Macaca mulatta (mono o macaco Rhesus) y Macaca fascicularis (M. cynomolgus, cinomolgo, macaco de cola larga o macaco cangrejero) son los más conocidos. El mono Rhesus (Macaca mulatta, Zimmerman, 1780) es natural de Asia y se distribuye de manera natural por Afganistán, China y Tailandia, desde el nivel del mar hasta los 3.000m de altitud. En cautividad o semilibertad, así como por reintroducción en hábitats nuevos, se puede encontrar por todo el mundo, y hasta hace poco era muy común en grupos en cautividad dependientes de gran número de laboratorios. Esta especie de mono es de 45 a 64cm de altura, entre 5 y 12 kg de peso, de hábitos sociales, que en libertad vive en grupos grandes, hasta de 50 individuos, con una expectativa de 35-40 años, pero se aceptan como seniles a los individuos que tienen más de 20 años y de «edad extremadamente avanzada» a los que llegan a la treintena13-15. La variación en la longevidad entre determinados individuos de las especies del género Macaca criados en centros de investigación, reservas o espacios protegidos (p. ej., los cayos y pequeñas islas del caribe) es muy acusada10,16. El macaco cangrejero (Macaca fascicularis; Raffles, 1821) es natural de Asia (Indochina, Filipinas, Indonesia) y vive hasta los 2.000m de altitud. En cautividad, supera los 35 años de vida. Es un macaco pequeño, de 40 a 65cm de longitud de cuerpo y con un peso que puede llegar hasta los 9 kg. En inglés se les conoce como long-tailed macaque. En estas dos especies se observa que los animales jóvenes (5 años) carecen de depósitos amiloides en sus cerebros, mientras que muchos de los animales viejos (mayores de 25-30 años) los presentan en la corteza cerebral17-19, aunque el grado de beta-amiloidosis y su implicación funcional sea muy discutible (véase sección 3). Se suele considerar que es mínima o inexistente la incidencia de neuropatología intraneuronal neurofibrilar relacionada con la proteína tau a esas edades19, pero que de forma constante presentan alteraciones cognoscitivo-comportamentales de diferente intensidad a partir de los 20-25 años, aunque diferentes autores describan distintos tipos de déficits tras el empleo de distintas técnicas para valorarlos y correlacionarlos con los observados en los humanos seniles normales y con EA19,20. Se puede afirmar que en la etapa senil existe siempre un cierto número de individuos (en proporción muy variable o imprecisa; véase sección 3) que presentan anomalías morfológicas y funcionales más profundas que la mayoría de la población13,17-20.
La proteína APP de los macacos Rhesus tiene una gran homología con la humana y una gran proporción de sus placas corticales es inmunorreactiva frente a anticuerpos selectivos contra Abeta40 humana, mucho más frecuentemente que frente a anticuerpos contra Abeta42 humano. En el 50% de las placas se detectó la enzima acetilcolinesterasa, con las mismas características bioquímicas que en las de los humanos; en el 20%, apolipoproteína E y en muchas de ellas, aunque en una pequeña proporción, existían proteoglucanos, heparina sulfato y alfa-quimiotripsina. Todos estos datos morfohistoquímicos son compartidos con la patología EA en humanos, pero se han encontrado importantes diferencias en la distribución y en la progresión espacial y temporal de las placas. Es decir, la distribución en estos animales difiere de la de los enfermos de EA, en donde es más marcada en hipocampo, amígdala, corteza entorrinal, córtex frontal y lóbulos temporal y parietal. La clásica progresión espaciotemporal de la patología descrita por Braak y Braak en el humano no parece que se dé en estas especies, aunque no exista publicado un estudio sistemático sobre el tema contando con una colonia de suficiente número de ejemplares en observación. Los macacos Rhesus con niveles moderados de Abeta presentan los primeros depósitos en zonas corticales de asociación. En animales con mayores cantidades de Abeta, estos depósitos se agrupan en áreas paralímbicas y en aquellos individuos con grandes cantidades de Abeta, a estas zonas se les suman áreas de la corteza límbica, auque el patrón es muy variable en estos casos. En algunos trabajos se ha descrito que la mayor concentración de placas se presenta en áreas frontales y en corteza somatosensorial primaria, mientras que las densidades menores se encuentran en amígdala, ínsula, córtex cingulado, región temporal límbica, corteza occipital y cortezas de asociación parietales17. La corteza motora y algunas áreas sensoriales parecen carecer de depósitos.
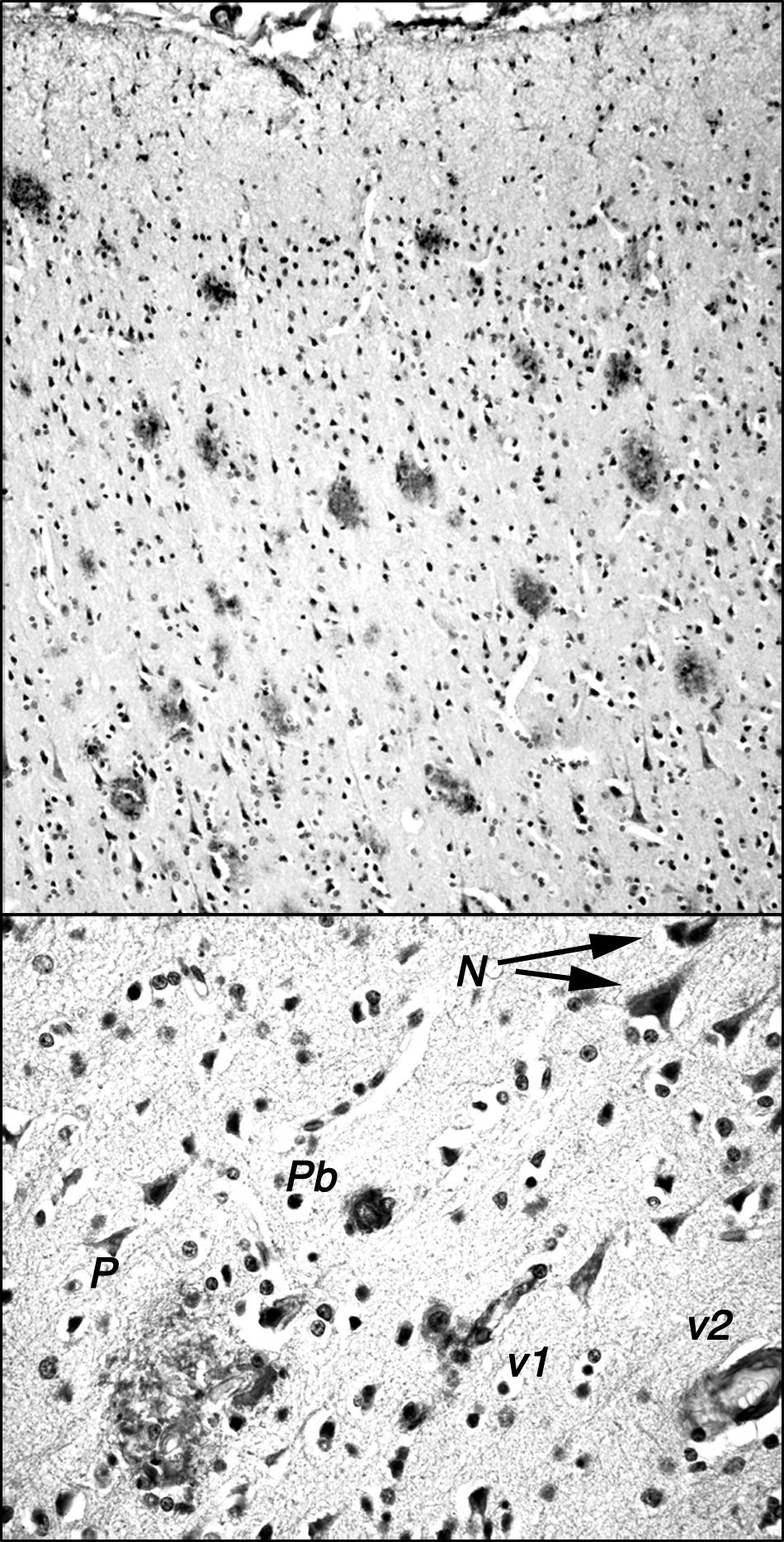
El macaco de cola de tirabuzón o rabón (Macaca arctoides; G. Saint-Hilarie, 1830) es natural de Asia (China, India, Malasia) y se encuentra hasta los 2.000m de altitud, pero existen colonias en algunas islas del Caribe y han sido criados también en cautividad o en semilibertad en bastantes laboratorios centroamericanos. Se estima su longevidad, en condiciones protegidas, en más de 30 años; tiene de 45 a 70cm de longitud del cuerpo y un peso entre 7 y 12 kg. En inglés se le conoce como stump-tailed macaque o bear macaque. En estos macacos de cola de tirabuzón, la situación involutiva morfofuncional cerebral parece ser similar a la de los Rhesus, aunque se aprecian algunas diferencias significativas. En la pequeña colonia de estos macacos que se estudia por este equipo de investigación en Cuba (fig. 1), se observa que a partir de los 24-26 años de edad existe una disminución de capacidades cognoscitivas-comportamentales objetivables mediante distintas pruebas neuropsicológicas (aprendizaje, realización de tareas) y observacionales (relaciones sociales, emotividad). Una prueba que ha aportado muy buenos resultados para determinar la involución funcional y su grado ha sido la prueba de demoras de aprendizaje-retención con muestras apareadas y desapareadas (delayed matching and nonmatching-to-sample [DMS y DNMS]), con ella se ha comprobado que ciertos individuos seniles del grupo presentaban un mayor déficit comportamental. En todos estos individuos se comprobó, a su muerte (entre los 25 y 34 años de edad), que existían alteraciones neuropatológicas relacionadas con la acumulación de amiloide. Se evidenciaron áreas de gran densidad de placas en diversas regiones cerebrales (corteza prefrontal, hipocampo, regiones parahipocampales) que coexistían con otras carentes de estas en esas regiones, así como placas dispersas en otras regiones del cerebro (fig. 2). Las placas son muy polimorfas y se presentan, además, depósitos difusos inmunorreactivos formados por agrupaciones de pequeñas áreas con inmunotinción homogénea o con muy finas granulaciones extracelulares inmunorreactivas, así como algunas granulaciones intraneuronales (fig. 2). Todos estos depósitos pueden ser visualizados con anticuerpos contra péptidos Abeta40 y/o Abeta42 humanos, aunque Abeta40 sea el principal componente tal como se ha descrito para los simios más evolucionados21. También, en algún caso, estamos observando pequeñas acumulaciones intraneuronales de proteínas reactivas con anticuerpos contra proteína tau fosforilada y no fosforilada humanas. Junto a estos animales con marcadas alteraciones neuropatológicas tipo Alzheimer, existen animales con alteraciones cognoscitivo-comportamentales menores y muy escasas alteraciones neuropatológicas. En ambos casos, las disminuciones neuronales fueron muy poco significativas frente a las cifras de densidad celular de adultos de menos de 20 años. No parece existir una intensa gliosis, salvo en algún ejemplar de los estudiados.
 de 34 años de edad, del Centro Internacional de Restauración Neurológica (CIREN), Cubanacán, La Habana, Cuba, estudiado en cautividad por los autores de la monografía. Tenía déficit cognoscitivo y comportamental muy acusado en los últimos meses de vida, significativamente superior a la media del grupo de su misma edad. Nótese el pelo blanquecino senil.")
Ejemplar de macaco de cola de tirabuzón (Macaca arctoides) de 34 años de edad, del Centro Internacional de Restauración Neurológica (CIREN), Cubanacán, La Habana, Cuba, estudiado en cautividad por los autores de la monografía. Tenía déficit cognoscitivo y comportamental muy acusado en los últimos meses de vida, significativamente superior a la media del grupo de su misma edad. Nótese el pelo blanquecino senil.
 de 34 años de edad, del Centro Internacional de Restauración Neurológica (CIREN), con déficit cognoscitivo-comportamental significativamente mayor que ejemplares «normales» de su misma edad. Las placas están marcadas con el anticuerpo 6E10 (Chemicon) que reconoce el epítopo 3-8 del péptido amiloide humano. En la parte superior se muestra la gran densidad de placas existentes en la corteza frontal, generalmente de tipo difuso no neurítico y sin core central, y con escasa gliosis. En la parte inferior, se muestra algunos otros depósitos amiloideos: N: acumulaciones granulares pequeñas intraneuronales; P: placa multiforme en la que se incluyen células de tipo glial y estructuras de muy diversa densidad de inmunorreacción; Pb: placa tipo burnt out de los enfermos de Alzheimer; v1: reacción amiloide adosada a un vaso de pequeño calibre y a las células que lo rodean; v2: reacción amiloide en las paredes de un vaso de mediano calibre.")
Placas amiloides en la corteza frontal de cerebro de un ejemplar de macaco de cola de tirabuzón (Macaca arctoides) de 34 años de edad, del Centro Internacional de Restauración Neurológica (CIREN), con déficit cognoscitivo-comportamental significativamente mayor que ejemplares «normales» de su misma edad. Las placas están marcadas con el anticuerpo 6E10 (Chemicon) que reconoce el epítopo 3-8 del péptido amiloide humano. En la parte superior se muestra la gran densidad de placas existentes en la corteza frontal, generalmente de tipo difuso no neurítico y sin core central, y con escasa gliosis. En la parte inferior, se muestra algunos otros depósitos amiloideos: N: acumulaciones granulares pequeñas intraneuronales; P: placa multiforme en la que se incluyen células de tipo glial y estructuras de muy diversa densidad de inmunorreacción; Pb: placa tipo burnt out de los enfermos de Alzheimer; v1: reacción amiloide adosada a un vaso de pequeño calibre y a las células que lo rodean; v2: reacción amiloide en las paredes de un vaso de mediano calibre.
Se han publicado trabajos estudiando las alteraciones consecutivas a la manipulación del cerebro de primates no humanos para dar lugar a animales con características «Alzheimer» (modelos de Alzheimer experimental22,23). Se han utilizado varios métodos, como lesiones microquirúrgicas o químicas en el córtex o los núcleos colinérgicos22-29, o microinyecciones de amiloide sintético o extraído de tejido de enfermos de EA30.Las diversas lesiones corticales y subcorticales en diversas especies han proporcionado bastantes adelantos en el conocimiento de la inervación colinérgica en el cerebro22-31 y su posible relación con la cascada amiloidea en la EA24. Sin embargo, no han permitido determinar con exactitud el papel que desempeña cada una de las estructuras corticales y subcorticales, acetilcolinotransmisoras y receptoras, en distintos comportamientos específicos de cada especie. En los simios del «nuevo mundo», las lesiones de los núcleos colinérgicos del cerebro basal inducían importantes daños en el aprendizaje o en las habilidades para memorizar, pero las correlaciones morfofuncionales se hacían difíciles de explicar. Esto se ha mostrado de manera muy evidente en marmosets (monos micos o titís de diferentes géneros de la familia cebidae, especialmente Callithrix, que tiene más de 18 especies)24,28,29. Así mismo, en un estudio utilizando monos ardilla (Saimiri sciureus), se inyectó ácido iboténico bilateralmente en el núcleo basal de Meynert y se originó destrucción de células en el putamen, globo pálido, estriado y amígdala, viéndose afectadas varias funciones cerebrales, como la discriminación visual de objetos, pero no otras, como la discriminación espacial. Sin embargo, el número de células dañadas no se correlacionaba con la cantidad de neurotoxina inyectada ni con el grado de pérdida de habilidades27. En contraste, las lesiones de núcleos basalocorticales de monos del «viejo mundo» no parecen producir déficits de memoria significativos ni duraderos, especialmente en macacos26. Esto puede deberse tanto a que en estos monos su cerebro más evolucionado tiene mayores capacidades de adaptación y circuitos neuronales más complejos y capaces de establecer alternativas frente a lesiones (al igual que sucede en el humano), como a las diferencias técnicas en los estudios con unos y otros primates (preentrenamiento en distintas tareas antes de la lesión, diferencias en los tests comportamentales, tiempo de inicio del estudio después de las lesiones, etc. 8,22,28-30). En cuanto a las diferencias entre los simios del nuevo mundo y el hombre, se podría pensar que las funciones cerebrales afectadas en el mono no son equivalentes a las humanas afectadas en la EA o bien que no se revelan con las pruebas que se emplean en el laboratorio. En los estudios de Ridley et al28,29, un período de 2 semanas después de la cirugía era suficiente para observar una recuperación de ciertas habilidades perdidas en primates, por lo que se sugirió que las modestas pérdidas de memoria observadas podrían deberse más a la pérdida de eferencias no colinérgicas del cerebro basal anterior al hipocampo y a otras estructuras límbicas relacionadas, que a la pérdida de la inervación colinérgica basalocortical a la corteza31. El conjunto de los resultados en modelos lesionales de primates no humanos aumenta la polémica sobre la teoría colinérgica de la EA porque no resuelve problemas sobre la base anatómica de las funciones cognoscitivas humanas22,23. Además, los resultados de estudios farmacológicos (usando una gran variedad de agonistas colinérgicos y reguladores enzimáticos) en estos animales no han producido éxitos como para fundamentar el uso de algún tratamiento para EA en humanos. En general, los tratamientos que han mejorado el sistema colinérgico en monos con lesiones en núcleos colinérgicos basales no han tenido efectos positivos en pacientes con EA22,25,29,31.
En 1998, se publicó que las microinyecciones estereotáxicas con Abeta40 sintética en la corteza frontal de macacos, especialmente si eran animales seniles, daban lugar a lesiones corticales dependientes de la dosis similares a las observadas en EA humana. Las zonas que rodeaban las lesiones presentaban, un corto espacio de tiempo después, neuronas distróficas, argirófilas y tioflavin-S fluorescentes, muchas de ellas inmunorreactivas frente a anticuerpos contra Alz-50 y ubiquitina32. Trabajos similares, realizados 5 años antes, habían fracasado en la demostración de la neurotoxicidad aguda de péptidos amiloideos en simios del nuevo mundo menos evolucionados (titís del género Callithrix)33. Sin embargo, con posterioridad, se demostró que microinyecciones intracerebrales de homogeneizados de cerebro de pacientes con EA en la fase media de la vida de los titís inducían, a los 6-7 años, la formación de placas de amiloide, neuritas distróficas argirófilas y angiopatía amiloide cerebrovascular34,35, a una edad en que nunca se observaba beta-amiloidosis. Sin embargo, en ninguno de todos estos primates se vieron ovillos neurofibrilares. Cabe resaltar que uno de los mayores objetivos de estos trabajos era aclarar si la «beta-amiloidosis» o «fibrilosis» de la EA era transmisible como lo parecían ser las beta-amiloidosis de origen priónico34,36,37. A uno de estos monos titís se le inyectó también intracerebralmente tejido de un paciente con una enfermedad priónica y se encontraron placas de amiloide y de angiopatía como en los cerebros de los monos inyectados con tejido de pacientes con EA34. Estos resultados, obtenidos con las herramientas de la década de los noventa, apoyaron la idea de que algunos casos de cualquiera de los tipos de beta-amiloidosis cerebrales, incluida la EA, pudieran ser inducidas por proteínas amiloides exógenas de configuración beta que, sin contener ácidos nucleicos, eran infectivas, considerando estos casos como enfermedades transmisibles34,35,38. Aunque actualmente la EA y las encefalopatías priónicas (denominadas encefalopatías espongiformes transmisibles) están firmemente separadas en este sentido, son muy grandes los lazos neuropatológicos y neuropatogénicos que las unen y que deben servir para progresar en nuestros conocimientos sobre estas enfermedades neurodegenerativas y lograr su tratamiento preventivo/curativo eficaz. La conclusión a que llegó el grupo de Ridley y Baker en los noventa era que «la beta-amiloidosis podía ser inducida por introducción de proteínas beta-amiloides»34,35, mientras que en 2000 manifestaban que «el beta-amiloide, o factores relacionados, pueden iniciar o acelerar el proceso de beta-amiloidosis cerebral en los primates»36. En la actualidad, se están generando modelos de Alzheimer en primates no humanos inyectando intracerebralmente Abeta42 unido a sustancias que bloquean la movilización del mismo, como el tiorfano30, tanto para estudios sobre la beta-amiloidosis como para ensayar nuevos fármacos.
Primates no humanos se han utilizado como modelo animal de elección en el ensayo de posibles fármacos o productos biotecnológicos («vacunas») útiles para el tratamiento de la EA, tanto preventivos como curativos; en el estudio de la posible influencia tóxico/neurodegenerativa de algunas sustancias o situaciones ambientales, y en la investigación de los mecanismos de acción de agentes neurodegenerativos o neuroprotectores. Todo ello con las cada vez más crecientes restricciones ya señaladas para el empleo de primates en experimentación.
Respecto a la utilización de primates no humanos en ensayos preclínicos de medicamentos para la EA, han sido muy importantes las aportaciones en estudios sobre medicamentos de tipo colinérgico y factores neurotróficos39-42, así como en el control de la amiloidosis43,44.
En el estudio de «vacunas contra el Alzheimer», los trabajos con cercopitecos han sido claves. Los macacos inyectados con péptidos amiloideos desarrollan altos títulos de anticuerpos anti-Abeta, con concentraciones en plasma 5 a 10 veces superiores a la de los animales control45,46. En un estudio con Chlorocebus aethiops inmunizados con péptido Abeta durante 10 meses47, en el día 42 los animales vacunados generaron en plasma grandes cantidades de anticuerpos anti-Abeta (2-5 veces superiores al control) que, además, marcaban placas humanas. También se encontraron anticuerpos en el líquido cefalorraquídeo pero con títulos menores, pero no se observaron respuesta de células T ni inflamación. En estos monos no parece que se presenten fenómenos secundarios importantes cuando se inmunizan, al contrario de lo que sucede en humanos. Es conocido que las vacunas con Abeta42 iniciaron el protocolo para una futura comercialización; sin embargo, no pasaron la fase clínica IIa en 2002 ya que 18 de los pacientes vacunados desarrollaron meningoencefalitis por activación de linfocitos T. Por ello se buscaron otras alternativas para los ensayos de las vacunas. Pero otros estudios posteriores realizados en macaco utilizando como antígeno el péptido Abeta15 mostraron resultados satisfactorios y seguros, incluida la no activación de linfocitos T: en la octava semana tras la vacunación se empezaban a desarrollar anticuerpos anti-Abeta42 en plasma e iban aumentando los títulos con las siguientes inoculaciones y disminuían después del período de vacunación. Además, este anticuerpo mostraba una alta especificidad contra Abeta4248. Los últimos resultados de los ensayos en primates hacen que algunos investigadores se muestren optimistas sobre las posibilidades de encontrar «vacunas» para la prevención y el tratamiento de EA y de que estas se puedan ensayar en los primates no humanos a pesar de la diferencia de reacción inmunológica entre las especies49. Asimismo los ensayos sobre anticuerpos contra monómeros u oligómeros amiloideos parece que pueden ser llevados a cabo en algunos primates no humanos con seguridad de que los resultados pueden ser extrapolados al hombre50.
Se han empleado primates no humanos en el estudio de la influencia de factores externos (medio ambiente, tóxicos, hábitos de vida) en el desarrollo de EA. Mencionaremos conclusiones de tres líneas de investigación que han tenido una gran repercusión: la restricción calórica atenúa la acumulación de amiloide, la exposición durante la infancia a algunos contaminantes ambientales puede predisponer a padecer EA y la presencia de ciertos metales en la dieta puede dar lugar a procesos neurodegenerativos.
En el primer caso se demostró que la restricción calórica hacía disminuir los depósitos amiloideos en el cerebro del mono ardilla (Saimiri sciureus), proceso que se demostraba relacionado con la disminución del estrés oxidativo51. También se demostró luego en macacos52.
En la segunda investigación se demostró que la exposición a plomo durante las primeras etapas del desarrollo inducía patología tipo Alzheimer en la senilidad53, confirmando estudios anteriores con roedores que indicaban que la exposición a este metal en etapas tempranas predeterminaba la expresión y la regulación del precursor de proteína amiloide y de la vía amiloidogénica54. En este estudio se trabajó con una cohorte de hembras de Macaca fascicularis divididas en dos grupos, uno control y otro al que se le inyectó acetato de plomo durante los primeros 400 días de vida. En 2003 se llegó a término con los animales, cuando ya tenían 23 años y se estudiaron varios de sus órganos, entre ellos el cerebro. En los animales seniles tratados con plomo, la expresión de los genes APP, BACE1 (β-site APP cleaving enzime1 o beta secretasa) y Sp1 (regulador transcripcional de los genes anteriores) estaba aumentada, así como los niveles de Abeta42 (100% de incremento). En cuanto a las alteraciones típicas de la EA, los cerebros de los animales expuestos a plomo mostraban un aumento de Abeta intracelular y de placas de núcleo denso. Igualmente se observaron placas difusas y algunos ovillos morfológicamente similares a los que se observan en humanos. La actividad de la ADN metiltransferasa 1 (DNMT1) estaba disminuida en un 20%, lo que hace pensar en que la expresión de estos genes estaba sometida a algún tipo de control epigenético mediado por mutilaciones. El biomarcador del daño oxidativo 8-oxo-guanina estaba también aumentado. Para explicar este fenómeno epigenético, los autores de este estudio proponen como hipótesis que los genes que están regulados por metilación pueden ser reprogramados en la edad adulta pues encontraron que 20 de los 22 genes que se alteraban por exposición a este metal son modificables por metilación. Esta idea sobre que los humanos puedan desarrollar EA tras la exposición a plomo durante el desarrollo está respaldada por el caso de un paciente que sobrevivió a la exposición severa de plomo cuando tenía 2 años, pero que falleció por un grave deterioro mental a los 42 años55. El cerebro de este paciente mostraba numerosas placas seniles y ovillos. Este trabajo justifica las suposiciones de que diversos agentes ambientales están implicados en el desarrollo de EA, así como la «hipótesis Barker» que asocia experiencias o agresiones tempranas en el curso de la existencia, donde se es especialmente vulnerable a estos agentes, a enfermedades de la edad adulta56,57.
Por último, varios trabajos han vuelto a señalar que algunos metales, como manganeso y aluminio, pueden inducir alteraciones de tipo neurofibrilar58.
DiscusiónEn la presente monografía se trata de contestar a la pregunta de si existe la EA en los primates, de manera general, o si este síndrome se produce exclusivamente en el hombre. Tal como se apuntó en la introducción, no existe posibilidad, en términos absolutos, de que ninguna especie padezca EA, pues ninguna posee las capacidades funcionales cerebrales superiores que configuran la mente del hombre (de memoria, conciencia, juicio, lenguaje, cálculo, etc.), pero en términos relativos sí cabe la posibilidad de que se produzcan alteraciones de las «funciones corticales superiores» en primates no humanos a causa de procesos degenerativos primarios del cerebro que se asemejen a la EA humana en patogenia y alteraciones de procesos cognoscitivos-comportamentales. Efectivamente, diversos autores llegan a la conclusión de que los (o algunos) primates no humanos ancianos desarrollan anormalidades acusadas, tanto comportamentales como morfológicas, en forma parecida a lo que ocurre en los humanos seniles y en los enfermos de EA14,59,60, pero esta conclusión merece diversas matizaciones no sólo por parte del tipo y significado de las anomalías, sino por el porcentaje de individuos que presentan esos cambios durante la senescencia. En este apartado se va a prestar especial atención a tres asuntos que pueden tener gran importancia para aumentar nuestros conocimientos sobre la EA: a) las similitudes y diferencias entre las lesiones neuropatológicas tipo Alzheimer de humanos y de primates no humanos; b) la prevalencia de las lesiones neuropatológicas y del posible «síndrome Alzheimer» en estas especies, y c) las implicaciones de las características involutivas-neurodegenerativas del cerebro de primates no humanos en la interpretación de las relaciones entre senilidad fisiológica e involución senil patológica (Alzheimer).
Similitudes y diferencias de las lesiones entre humanos y primates no humanosEstudios llevados a cabo en una gran variedad de especies de primates no humanos, desde los menos evolucionadas hasta los más cercanos al hombre, no solo no aclaran el porqué de la existencia de la patología amiloidea y de la patología relacionada con la proteína tau, sino que plantean más interrogantes respecto a las teorías sobre la neurodegeneración y la EA. De una manera genérica, se puede decir que, en los primates, se genera durante la senilidad una marcada neuropatología relacionada con la acumulación de amiloide, aunque no en todas las especies y con afectación de un número muy variable de individuos de su población, mientras que sólo excepcionalmente (casi exclusivamente en el hombre) aparece una clara neuropatología relacionada con la proteína tau (neurofibrilosis, taupatía o tauopatía). Por qué la beta-amiloidosis aparece siempre en la senilidad de algunas especies (cercopitecos) y no en otras (algunos tamarinos) y, por otro lado, por qué la beta-amiloidosis se asocia a la neurodegeneración del cerebro en algunos individuos de una especie (macacos), tal como ocurre en la EA del humano, respetando a otros «seniles normales», no es fácil de entender. Tampoco lo es la errática incidencia, aunque siempre de escasa intensidad salvo en el hombre, de neuropatología relacionada con la proteína tau en las diversas especies de primates.
La APP es una proteína ancestral de membrana que aparece en seres unicelulares y se expresa abundantemente en cerebros de mamíferos. Es una proteína que se conserva con muy pocas variaciones a lo largo de la evolución, existiendo su doble degradación «amiloidogénica» y «no amiloidogénica» en todos los mamíferos5,61,62. Sin embargo, la beta-amiloidosis no se presenta en la evolución de los mamíferos hasta los primates (salvo en muy raros casos —osos, perros—, ya mencionados en la primera parte1). En el caso del «mono musaraña», Tupaia belangeri, pro-primate del orden Scandentia, muy anterior al orden Primate y más próximo a los roedores1, cuya secuencia del péptido beta amiloide es idéntica a la de los humanos, no se ha evidenciado ningún tipo de beta-amiloidosis interneuronal ni asociada a los vasos sanguíneos63. En el orden Primate, parece existir ya de manera clara y bastante constante la neuropatología amiloidea, siempre relacionada con la edad avanzada, aunque no se correlacione la incidencia, ni la prevalencia, ni la intensidad de la beta-amiloidosis con la situación de cada género en la escala evolutiva dentro de este orden, ni con la presencia o no de alteraciones cognoscitivo-comportamentales. Todo esto puede hacer suponer que en los mamíferos existe una «resistencia» innata a la beta-amiloidosis cerebral, que por una parte se debe a que la secuencia de aminoácidos del péptido amiloide es una en mamíferos que pueden producir beta-amiloidosis (primates, perros, osos) y otra en los que no (roedores, rumiantes)36, y por otra parte, se facilita la eliminación de productos de configuración beta e insolubles. Sin embargo, en determinadas circunstancias, con la edad y en situaciones no bien conocidas, se alteran los factores que regulan esa producción y/o eliminación de productos amiloideos61 en algunos primates, los mamíferos más evolucionados. Podemos suponer que las situaciones desencadenantes de beta-amiloidosis son las propuestas para el desarrollo del Alzheimer humano: un marcado estrés oxidativo, reacciones neuroinflamatorias, inducción de apoptosis, etc.
Los tipos morfológicos y morfohistoquímicos de lesiones amiloideas presentes en los primates no humanos se corresponden solo en parte con los que se describen en humanos1,9,17-19,64-66], en donde ya existen muchas variedades, lo que es difícil de explicar sin recurrir a la existencia de una gran heterogeneidad molecular en los depósitos67. En el apartado 4.5 se mostraba cómo en lémures y macacos existían distintos tipos de depósitos: placas parenquimatosas, depósitos difusos (no en placas) y depósitos perivasculares. Las acumulaciones en placa presentan formas e inmunorreacción (tanto frente a amiloides Abeta40 y Abeta42 como a otros componentes, como neprilisina68) mucho más variables que en el hombre, lo que no ayuda a corroborar ni a negar las teorías que algunos autores han propuesto para explicar la génesis y la evolución de estas estructuras en humanos. Los depósitos difusos de amiloide sí se correlacionan mejor con los que se observan en humanos, tanto en la corteza como en zonas subcorticales, aunque están muy poco estudiados, tal como ocurre en el hombre. Los depósitos vasculares parecen ser muy abundantes en los simios menos evolucionados, formando cintas que rodean los vasos, pero son más escasos en los antropoides y el hombre. Además, como ya se mencionó, la evolución espacio-temporal de la patología amiloidea no se cumple en los primates no humanos. Esto pudiera ser un gran handicap para considerar estos primates como modelo de Alzheimer, aunque en algún caso se haya correlacionado bien la carga amiloidea con la atrofia cerebral69,70.
Uno de los grandes «inconvenientes» que se ha encontrado en los primates no humanos como modelos de Alzheimer ha sido su escasa, por no decir nula, neuropatología relacionada con la proteína tau en la forma en que se observa en los cerebros de humanos que padecieron EA. Es decir, en simios y prosimios hay una práctica inexistencia de ovillos neurofibrilares y neuritas distróficas, así como muy escasa inmunorreactividad neuronal cuando se emplean anticuerpos contra proteína tau y, especialmente, tau altamente fosforilada. Esto podría interpretarse en el sentido de que estas beta-amiloidosis de los primates no humanos serían «otras patologías» distintas del Alzheimer humano. Sin embargo, algunos de los trabajos publicados en los últimos años parecen contradecir esta interpretación. Sin entrar en la controversia de si la EA es esencialmente una beta-amiloidosis o una taupatía (o tauopatía), ya que se puede considerar que ambos procesos patológicos son consustanciales a la EA, aunque con diferente incidencia en distintos enfermos, son dos los aspectos que se deben tener en cuenta. En primer lugar, bajo la denominación de taupatía o tauopatía se agrupa actualmente toda la serie de enfermedades o procesos neurodegenerativos (EA, Parkinson, etc.) en donde está implicada la proteína tau, sea cual sea su grado y forma de afectación (en somas, dendritas o axones; formando acumulaciones o sin hacerlo, siempre que induzca alteraciones estructuras neuronales), así como su correlación con otras alteraciones propias de cada enfermedad, tal como sucede en distintos casos de EA que presentan una baja o alta incidencia de neuropatología tau. En segundo lugar, se han publicado varios trabajos en los que se pone de manifiesto, empleando técnicas de mayor sensibilidad (al microscopio óptico o al microscopio electrónico; usando técnicas histoquímicas de mayor resolución), que se observan alteraciones morfohistoquímicas relacionadas con la proteína tau (microacumulaciones de depósitos, con mayor o menor fosforilización, en algunos somas neuronales o neuritas distróficas, o bien relacionados con sinapsis alteradas o con depósitos amiloides)71-73. Incluso se ha publicado un caso de chimpancé en el que se detectaron filamentos helicoidales apareados66.
Incidencia y prevalencia de «neuropatología Alzheimer» y de un posible «síndrome Alzheimer» en las distintas especies de primates no humanosExisten especies de primates no humanos, pro-simios y simios, en los que la presencia de depósitos amiloideos en los animales seniles es bastante constante. El ejemplo más claro es el del simio Chlorocebus aethiops, el cercopiteco verde, en donde el 100% de los animales seniles presentan amiloide. En estos animales, aunque carecemos de estudios congnoscitivo-comportamentales en profundidad, parece que la beta-amiloidosis no se acompaña de graves alteraciones de las funciones cerebrales que pudiéramos interpretar como neurodegeneración cerebral.
Frente a ello, se observa que los macacos (Macaca mulatta y M. fascicularis), modelos selectivos de primates no humanos para muchos autores, que están además filogenéticamente muy próximos al género Chlorocebus, dentro de la misma familia Cercopithecoidea, manifiestan una generalizada, de leve a moderada, involución cerebral en la senilidad, en la que sólo algunos individuos presentan anomalías morfológicas y funcionales más profundas que la mayoría de la población cuando envejecen13,17-19. Estos últimos individuos podrían sufrir un «síndrome Alzheimer». Sin embargo, se puede cuestionar esta conclusión por varios motivos, especialmente por la indefinición de la prevalencia de la neuropatología Alzheimer y de la naturaleza y las características de las alteraciones cognoscitivo-comportamentales. El porcentaje de individuos más afectados en esta especie no se ha podido establecer, ni siquiera de forma aproximada, ya que es muy escaso el número de ejemplares que comprende cada estudio. Solo existe en observación una gran colonia de estos monos en libertad en la isla de Cayo Santiago, cercana a las costas de Puerto Rico, dependiente del Centro Caribeño de Investigación de Primates de la Universidad de Puerto Rico. En ella hay más de un millar de individuos, aunque distribuidos en varias subcolonias familiares16, pero con ellos no se desarrollan proyectos de investigación sobre involución/neurodegeneración cerebral. Los datos que sobre este tema aportan las publicaciones mencionadas anteriormente son muy contradictorios. Inicialmente, autores muy relacionados con el estudio de la EA encontraron anomalías muy profundas en los macacos ancianos, como intensas pérdidas neuronales y sinápticas en corteza prefrontal e hipocampo y beta-amiloidosis9,17-19,74,75. Posteriormente, otros estudios mostraron que las alteraciones morfohistoquímicas no eran tan marcadas, incluso que las pérdidas neuronales eran escasas aunque existieran déficits cognoscitivos importantes76-78. Estos estudios incluso dan pie a cuestionar si el síndrome de Alzheimer en el humano no es un proceso patológico previo y/o parcialmente independiente de la beta-amiloidosis.
Estudios puntuales sobre aspectos muy concretos que pueden ser interpretados como marcadores de la patología Alzheimer en macaco Rhesus (acumulación de distintos tipos de péptidos amiloideos y proteína tau —muy escasa ésta última—; respuestas a determinados tests cognoscitivo-comportamentales) pueden ofrecer resultados de porcentajes de afectación entre el 0 y el 100% en los diferentes grupos de animales estudiados17-19. La mayor prevalencia de acumulaciones amiloideas (casi en el 100% de los individuos analizados) en monos Rhesus ancianos, de 25 a 30 años de edad, es la descrita por Satu et al en 200317. Pero solo en casos muy concretos se puso de manifiesto que había macacos seniles con marcados depósitos amiloideos, acusadas alteraciones en el núcleo basalis y trastornos profundos del comportamiento y del aprendizaje (que podríamos denominar como indiciarios de un síndrome Alzheimer)13. Los estudios llevados a cabo en otros macacos, como en el macaco cinomolgo o macaco cangrejero (Macaca fascicularis) y en el macaco de cola de tirabuzón (Macaca arctoides), proporcionan resultados muy parecidos: aparece intensa patología amiloidea tipo Alzheimer en un porcentaje variable, aunque estimamos que no es superior al 20 o 30%, de individuos seniles, sin que se pueda precisar la prevalencia en la especie debido al corto número de los ejemplares en estudio17,18, aunque sí se constata que la mayoría de ellos presenta déficits cognoscitivos-comportamentales muy profundos asociados a estas intensas manifestaciones neuropatológicas. Frente a esto, se han encontrado macacos seniles «normales» con alteraciones escasas tanto del comportamiento social y del aprendizaje como de la estructura cerebral20.
En cuanto al significado de los déficits cognoscitivos-comportamentales, se esgrimen distintas opiniones según se consideren las relaciones que existen entre las «funciones superiores» propias de cada especie y/o las pruebas neuropsicológicas y de comportamiento que han servido para evaluar esas funciones en cada especies79,80. Cuando se quiere estudiar la estructura y la función del cerebro en cada especie o el desarrollo evolutivo global, estas cuestiones son de radical importancia, pero no tienen tanto interés en el estudio de la involución cerebral de cada especie. En ella es más importante evaluar la pérdida de funciones de vital importancia para la supervivencia del individuo (aprendizaje, estrategias para alimentarse o sortear peligros, etc.) y el mantenimiento de sus relaciones sociales81. Todas estas son las que están muy alteradas en los animales que se pueden considerar como afectados por el «síndrome Alzheimer»7. En una publicación del año 2007, resultado de un simposio sobre envejecimiento cerebral, se analizaron las características de la involución senil de los macacos y se llegó a la conclusión de que, al igual que ocurría en los humanos, existía un «exitoso» y un «no exitoso» proceso de envejecimiento en el que no aparecían o sí aparecían déficits cognoscitivo-comportamentales muy estadísticamente significativos frente a la media en cada grupo de edad82.
En su conjunto, los estudios ponen de manifiesto la posible existencia de un síndrome EA en macacos, pero sin concretar sobre su prevalencia y su similitud con la EA en humanos, mientras que los monos verdes del género Chlorocebus parecen indicar la existencia de un posible envejecimiento con alteraciones neuropatológicas tipo Alzheimer sin grave deterioro cognoscitivo tipo Alzheimer. Las acumulaciones amiloideas parecen ser menos neurotóxicas cuando se producen con gran incidencia y prevalencia en los animales seniles, caso del C. aethiops, donde no se han descrito grandes problemas cognoscitivos-comportamentales en los animales ancianos. Pero cuando se presentan solo en una reducida «subpoblación» de una especie (que podemos considerar que padece una «involución patológica senil tipo Alzheimer»), como sucede en el hombre y en los macacos, los daños en sinapsis, neuronas y circuitos neuronales son muy importantes y condicionan graves trastornos mentales y comportamentales. Las propiedades neurotóxicas de los depósitos beta-amiloideos pueden ser diferentes en las distintas especies de primates, incluido el hombre, sobre la base de su distinta composición y organización macromolecular. Cuando se agregan los monómeros y oligómeros de beta-amiloide para formar fibrillas y depósitos insolubles, procesos no bien conocidos todavía, pueden originarse distintos tipos fisiopatológicos de acumulaciones, siendo uno de ellos característico (y etiopatogénico) del Alzheimer humano y otros no. Esta concepción viene avalada por el hecho de que algunas sustancias presentan diferente reactividad con los depósitos amiloideos humanos y los no humanos, siendo de especial interés la gran reactividad del compuesto PIB (benzothiazole imaging agent Pittsburgh Compound B, usado para marcar placas en estudios de tomografía computarizada por emisión de fotones simples) en humanos, que no se observa en otros primates (mono ardilla, macacos)83.
Involución senil fisiológica e involución senil patológica (Alzheimer). El «continuum» entre «senilidad» normal y enfermedad de AlzheimerLa alta variabilidad en la existencia de neuropatología Alzheimer entre especies de primates no humanos y entre diferentes individuos de una especie tiene también importantes implicaciones en el desarrollo de las teorías que intentan explicar la relación entre la senilidad fisiológica y la EA. Chlorocebus aethiops (con el 100% de los animales seniles presentando depósitos de amiloide, con alteraciones comportamentales no intensas) y Macaca mulatta y M. fascicularis (con muy variable grado de amiloidosis, hasta el 100% en algún grupo de estudio pero quizás no superior al 30% de los individuos seniles de estas dos especies, pero sí incluyendo en esta subpoblación animales con profundas anomalías cognoscitivo/comportamentales), todos ellos de la familia Cercopithecoidea, nos indican que existen patrones de involución senil muy diferentes, tanto propios de género y especie como de cada individuo, en donde la amiloidosis cerebral tiene una implicación variable tanto en el proceso de envejecimiento «normal» o «con no profundas alteraciones cognoscitivo/comportamentales», como en la neurodegeneración, «envejecimiento patológico» o «envejecimiento tipo Alzheimer». Esto nos hace pensar que no conocemos bien todavía las implicaciones cognoscitivo/comportamentales de la beta-amiloidosis cerebral.
La concepción que de la EA tienen diversos autores se sitúa en dos polos prácticamente opuestos: a) la EA es una enfermedad neurodegenerativa ligada a la edad, en su forma esporádica, que se desarrolla en ciertos individuos predispuestos genéticamente por la acción de otros factores intrínsecos (diabetes, problemas vasculares, etc.) y factores extrínsecos (poco conocidos), siendo totalmente diferenciable de la senilidad, y b) la EA es la última fase de la involución senil del cerebro. En este último sentido, Alzheimer llegó ya a considerar que el caso clínico que describía era una manifestación de envejecimiento intenso y acelerado del cerebro, y muchos estudios epidemiológicos y evolutivos modernos han calculado que, a una edad aproximada de 110 años, la EA llegaría a tener un 100% de prevalencia, siendo precisamente los genes responsables de la evolución del hombre y del desarrollo de las regiones más evolucionadas del cerebro los responsables de la aparición de la EA83,84. Diversos expertos consideran que existe una transición gradual («continuum») desde la senilidad normal fisiológica hasta las últimas fases del Alzheimer, pasando por «la alteración o deterioro cognitivo leve» (ACL o DCL, equivalente al mild cognitive impairement [MCI]), mientras que otros expertos piensan que existe una involución individualizada que conduce a diferentes situaciones85,86. La involución funcional cerebral en cada individuo tiene características propias y tanto la EA como la ACL/DCL/MCI aparecen a edad variable, de los 60 a los 100 años y con una intensidad en lesiones y manifestaciones neuropatológicas que no se correlacionan con la edad85-87. Podrían pues existir a determinadas edades diferentes poblaciones de «ancianos normales», «ancianos normales con —ligeras— alteraciones neuropatológicas» y enfermos de ACL/DCL/MCI y de EA con diferente deterioro clínico y anatomopatológico que podrían ser «subpoblaciones» de diversos procesos de envejecimiento o bien de un único proceso de envejecimiento continuado del cerebro, incluso individuos muy ancianos con un «exitoso» envejecimiento en el plano funcional, aunque presenten alteraciones neuropatológicas cuando son estudiados sus cerebros87.
Los estudios de prevalencia de la EA según la edad, la constatación de que una parte del grupo ACL/DCL/MCI desarrolla EA en un plazo de 4-5 años85,86 y la característica de progresividad espacio-temporal clínico-patológica de la EA son, para algunos autores, pruebas concluyentes de que existe un claro «continuum» senilidad fisiológica-EA. Pero ellas podrían ser sólo indicativas de la progresividad de la EA y la aparentemente mayor facilidad para desarrollarse en edades cada vez más avanzadas del humano. La subpoblación humana de ancianos no dementes, incluso con alteraciones ligeras-moderadas neuropatológicas, podría equivaler a la gran subpoblación de cercopitecos seniles con escasos daños comportamentales. En este sentido, esta subpoblación, aunque en su aspecto negativo fuera el punto de partida para un proceso neurodegenerativo EA, tendría un gran interés teórico pues podría significar también la posible existencia en la involución senil fisiológica de una fase estable sin demencia. Investigaciones multidisciplinarias deberían llevarse a cabo para lograr estabilizar la involución senil humana en esta fase.
La causa última de este proceso involutivo senil especial del cerebro en el humano habría que buscarla en los cambios genéticos ocurridos durante la evolución83,84,88-92. Diversos genes fueron seleccionados, y diversas mutaciones de los mismos, para llegar a los primates no humanos y, finalmente, al hombre. La adaptación al medio ambiente ancestral con una vida más corta de duración desarrolló un genoma óptimo en esos momentos pero que en las actuales situaciones y con una expectativa de vida mucho más larga, puede ser fuente de cambios patológicos muy importantes en el final de la vida. En este sentido, el paso evolutivo a los primates parece iniciar las condiciones para que se desarrolle una nueva enfermedad degenerativa ligada a la edad, que luego tiene su manifestación más florida en la especie humana. Las distintas manifestaciones neuropatológicas (amiloidosis, alteraciones tau dependientes, estrés oxidativo, etc.) entre los distintos géneros y especies de primates, incluido el humano, son el reflejo de una distinta evolución de los genes de las proteínas APP y tau, con enzimas relacionadas con su metabolismo, y con enzimas respiratorias mitocondriales88-92.
ConclusionesConsiderados en su conjunto, los primates no humanos (verdaderamente) seniles presentan grandes variaciones (tanto entre los géneros y las especies como entre grupos o individuos de una determinada especie) en déficits cognoscitivo-comportamentales y en la prevalencia y en la incidencia y tipo de alteraciones neuropatológicas (esencialmente amiloideas) similares a los que se observan en los humanos que padecen o padecieron EA. Alteraciones neuropatológicas parecidas a las de la EA humana, y raramente vistas en otros mamíferos (en su gran mayoría beta-amiloideas, pues sólo en algunos casos se detectan acumulaciones inmunorreactivas a proteína tau fosforilada), se observan en todos los subórdenes evolutivos de los primates, desde los prosimios (lémures) a los grandes simios (chimpancé), pasando por los platirrinos (cebús, monos ardilla) y catarrinos no hominoides (macacos y cercopitecos). Las disminuciones de las funciones cognoscitivo-comportamentales no están bien documentadas, pero parecen existir en casi todos los animales estudiados realmente seniles, aunque sólo de forma intensa en algunas especies. Tras el análisis de los resultados publicados y de nuestras observaciones en el estudio comportamental y neuropatológico de una colonia de macacos (Macaca fascicularis, macaco cangrejero), se puede llegar a la conclusión de que en algunas especies de primates no humanos existen individuos en los que se presenta un síndrome neurodegenerativo muy acusado que se asemeja a la EA humana frente a una involución senil muy poco marcada por alteraciones neuropatológicas y déficits cognoscitivo-comportamentales de congéneres de su misma edad. Sin embargo, esta conclusión no es extrapolable a todos los primates no humanos, pues parece que en otras especies, incluso muy próximas en el desarrollo evolutivo (como ejemplo, la especie Chlorocebus aethiops, mono o cercopiteco verde, de la misma familia que los macacos), se pueden presentar alteraciones neuropatológicas de forma constante y escasos déficits cognoscitivos, o bien pocas alteraciones de todo tipo en otras especies. La prevalencia de las alteraciones neuropatológicas más graves observables en las distintas especies es muy variable: desde un porcentaje del 100% en Chlorocebus a un porcentaje inferior al 30% en los macacos. Las diferencias entre senilidad normal y patológica (Alzheimer) son difíciles de establecer por la falta de estudios cognoscitivo-comportamentales en muchos grupos analizados, así como por la controversia existente en estos estudios cuando se llevaron a cabo (en metodología e interpretación de datos), aunque en algunos casos concretos, en macacos especialmente, se haya comprobado la citada correlación entre un alto grado de deterioro funcional cerebral y una gran cantidad de alteraciones neuropatológicas (posible patología Alzheimer). En algunos casos (macacos), se aprecia la existencia de un posible «continuum» entre proceso de envejecimiento «normal», «normal con no profundas alteraciones neuropatológicas y cognoscitivo-comportamentales» y «envejecimiento patológico» o «envejecimiento tipo Alzheimer». En otros casos, las alteraciones neuropatológicas son constantes y bastante marcadas, pero sus repercusiones cognoscitivo-comportamentales no parecen muy importantes (cercopitecos verdes); ello hace suponer la posible existencia en la involución senil fisiológica de una fase estable sin demencia, aun cuando existan alteraciones neuropatológicas. El estudio de estas patologías seniles primarias en los primates no humanos, aun cuando existan dificultades por la longevidad de los individuos en algunas especies y por problemas legales en el trabajo con primates, permite extraer importantes conclusiones que pueden ser de aplicación en el campo de la EA humana, ayudando a explicar e interpretar la patogenia de este síndrome y a descubrir posibles dianas terapéuticas.
FinanciaciónEl trabajo ha sido financiado en su mayor parte con fondos propios de los laboratorios implicados, y en el periodo 2010-2011, por una ayuda del Plan Nacional (CTQ 2009-09538)
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

