



Transthyretin-related familial amyloid polyneuropathy (TTR-FAP) typically arises as an autonomic neuropathy primarily affecting small fibres and it occurs in adult patients in their second or third decades of life. It progresses rapidly and can lead to death in approximately 10 years. Other phenotypes have been described in non-endemic areas.
Objectives and methodsWe described 4 cases from the Spanish province of Guipuzcoa, a non-endemic area, to highlight the clinical variability of this disease.
Patients and resultsThree patients presented a late-onset form manifesting after the age of 50, featuring a predominantly motor polyneuropathy initially causing distal impairment of the lower limbs followed by the upper limbs. One patient suffered severe neuropathic pain. None showed signs of autonomic involvement. The fourth patient, of Portuguese descent, presented a typical form with onset in her thirties, neuropathic pain and dysautonomia. All patients carry the Val50Met mutation in the TTR gene.
ConclusionFAP is a pleomorphic disease even in patients carrying the same mutation. In non-endemic areas, its main form of presentation may resemble a predominantly motor polyneuropathy developing in the sixth decade of life with no signs of dysautonomia. Given this non-specific presentation and the widely available technical means of studying the TTR gene, we believe that the protocol for the aetiological diagnosis of any polyneuropathy should include genetic sequencing of TTR.
La polineuropatía amiloidótica familiar por transtiretina (PAF por TTR) cursa inicialmente con una afectación preferente de fibra fina e importante afectación autonómica, con un inicio en la segunda-tercera década de la vida y una evolución rápidamente progresiva que puede llevar a la muerte del paciente en unos 10 años. Sin embargo, en zonas no endémicas, se han descrito otros fenotipos.
Objetivos y métodosDescribimos 4 casos procedentes de una zona no endémica, la provincia de Guipúzcoa, con el objetivo de mostrar la variabilidad clínica de esta enfermedad.
Pacientes y resultadosTres pacientes presentaron una forma de inicio tardío, por encima de los 50 años, que cursó como una polineuropatía de predominio motor con afectación distal inicial en miembros inferiores y posteriormente en los superiores. Uno sufrió dolor neuropático intenso. Ninguno tuvo signos de afectación autonómica. La cuarta paciente, de origen portugués, presentó una forma típica de inicio en la treintena, con dolor neuropático y disautonomía. Los 4 pacientes presentan la mutación Val50Met en el gen TTR.
ConclusiónLa PAF es una enfermedad pleomórfica incluso en pacientes con la misma mutación. En zonas no endémicas, su presentación predominante puede ser como una polineuropatía de inicio por encima de la sexta década, de predominio motor y sin signos disautonómicos. Dado lo inespecífico de esta forma de presentación y la facilidad técnica con la que se puede estudiar actualmente el gen TTR, creemos que en el protocolo de diagnóstico etiológico de cualquier polineuropatía se debe incluir la secuenciación de este gen.
Transthyretin familial amyloid polyneuropathy (TTR-FAP) is a genetic, multisystem disease caused by mutation of the TTR gene.1–7 Corinho Andrade first described the condition in 1952 in northern Portugal,2,3,8 where it is colloquially referred to as mal dos pezinhos.5
The disease is most frequent in 3 endemic foci, in Portugal, Sweden, and Japan.2,4,5,8,9 The most significant foci in Spain are located on the island of Mallorca7–9 and in Valverde del Camino (province of Huelva).7 The Basque Country is a non-endemic area. Prevalence of TTR-FAP is estimated at 1/10000 population globally1 and 1/100000 population in Europe.7,8
TTR is a short gene, containing only 4 exons, located on the short arm of chromosome 18 (18q12.1).5 Over 100 mutations have been described in association with the gene. The most frequent mutation is a valine-to-methionine substitution at position 30 (Val30Met or V30M, now denominated Val50Met due to an update to the reference sequence entailing a change in the order of amino acids) of the 127 amino acid sequence.1–8
Transmission mainly follows an autosomal dominant pattern of inheritance,7,8 although sporadic and de novo cases have also been reported.5 Penetrance is highly variable, and differs according to the mutation and endemic focus.5–8 Early symptom onset and increased penetrance have been associated with maternal transmission of the mutation.5,8
TTR is an amyloid precursor protein which transports thyroxine and vitamin A.1,5–8 It is mainly synthesised in the liver (95%), but is also expressed in the retina and choroid plexuses. The protein circulates as a tetramer in the blood and the cerebrospinal fluid. TTR mutations destabilise the tetramer, giving rise to abnormally folded monomers which are distributed patchily in various tissues, especially in the peripheral nervous system and the heart.1,5–7
The variability in the age of symptom onset and the clinical presentation of TTR-FAP, especially in non-endemic areas, makes diagnosing the condition a complex, lengthy process.
Patients, methods, and resultsWe describe the clinical variability of TTR-FAP as observed in 4 identified patients consulting at the Hospital Universitario Donostia neurology department between 2005 and 2016.
Patient 1Patient 1 was a 58-year-old man from Navarre who attended the neurology department in December 2013 due to a 1-year history of fatigue and a “strange” sensation in the feet. In recent months, he had experienced a similar sensation in the hands, hindering certain fine movements, such as opening a clothes peg. He displayed no signs of dysautonomia.
The patient had no relevant history except left-sided amblyopia since infancy and surgery for retinal detachment and cataracts. The neurological examination identified no abnormalities, except mild weakness in dorsiflexion of the ankles and a mild sensory alteration in a “stocking” pattern.
The complete blood count and urine sediment test (including microalbumin determination) yielded normal results. An electromyography study of the short thumb abductor and the left tibialis anterior muscle showed no alterations. Electroneurography findings were compatible with axonal sensorimotor polyneuropathy with predominance of sensory alterations. A sural nerve biopsy revealed a severe axonal neuropathy with no amyloid deposition and no sign of vasculitis.
Both motor and sensory symptoms worsened progressively. In August 2015, the patient's uncle was diagnosed with an amyloid neuropathy after a sural nerve biopsy. This led us to order sequencing of the TTR gene, which revealed the c.148G>A (p.Val50Met) mutation in heterozygosis. An echocardiogram revealed amyloid deposition on the interventricular septum and mild diastolic dysfunction. An ophthalmological examination detected no amyloid deposition. Treatment was started with triflusal and bisoprolol.
We performed a genetic study of the family to identify potential carriers of the mutation. The patient's aunt and brother, aged 87 and 68, respectively, carried the Val50Met mutation, but presented no symptoms or alterations.
Patient 2Patient 2 was a 77-year-old man from Gipuzkoa who attended the department complaining of a 2-year history of fatigue, pain, and a tingling sensation in the feet, interfering with his ability to walk; he also reported a tingling sensation in the fingers. He displayed no signs of dysautonomia.
He had a history of arterial hypertension (treated with indapamide) and constipation (treated with isphagula husk). His parents had died at the ages of 96 and 92, with no neurological diseases. He had 4 older siblings (aged 87, 84, 82, and 81), with no relevant family history of neurological problems.
The neurological examination revealed paralysis of the distal muscles of the legs; amyotrophy of the thenar eminence and the interosseous muscles of the hands; reduced touch, pain, and vibration sensation in a “stocking” distribution and in the fingers; universal areflexia; and mildly ataxic gait with bilateral steppage.
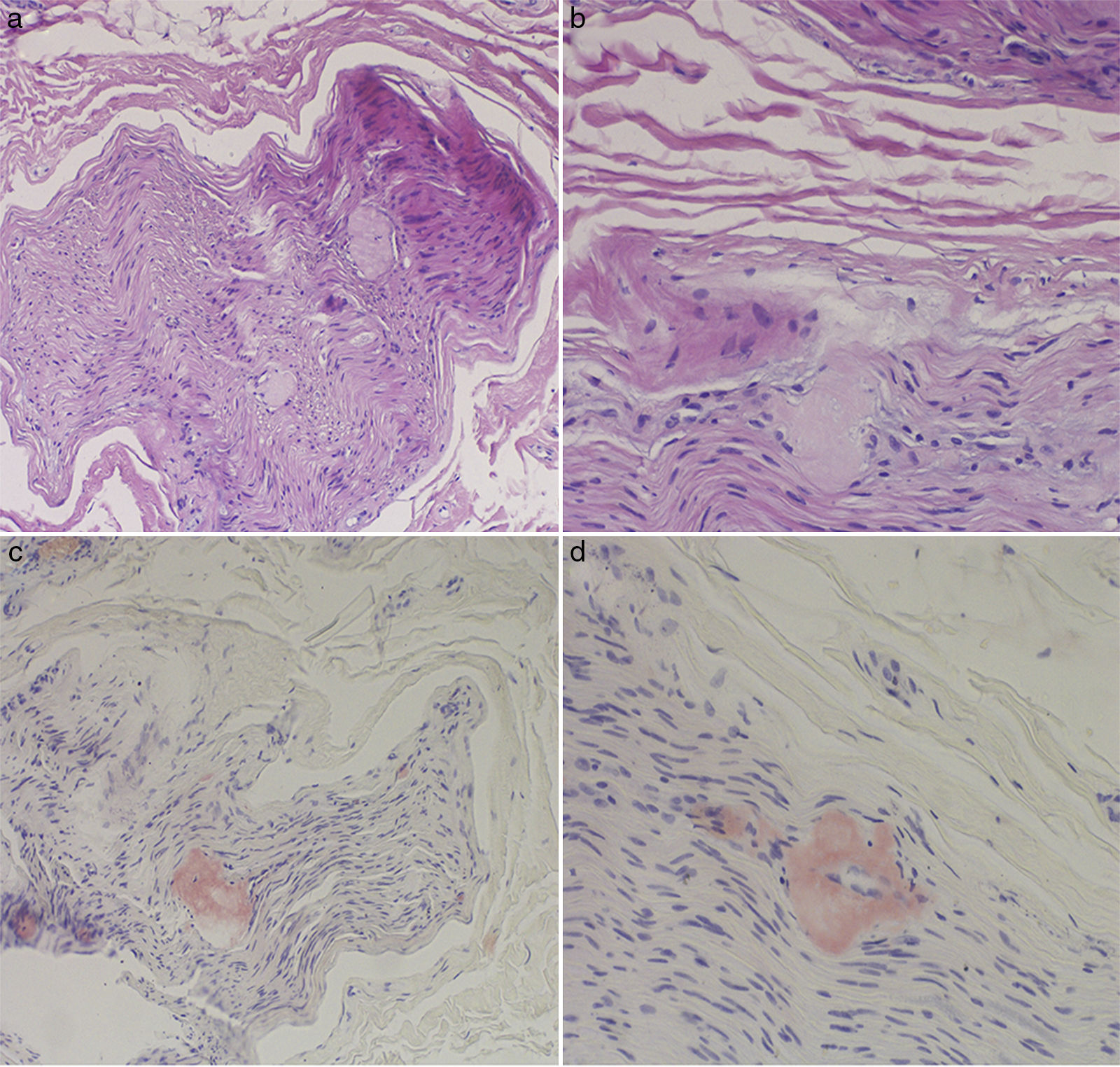
The complete blood count and urine sediment test yielded normal results. Electroneurography findings were compatible with severe axonal sensorimotor polyneuropathy with predominance of sensory alterations. A sural nerve biopsy detected amyloid deposits destroying the nerve fibres; the deposits were positive for Congo red staining (Fig. 1).
 and Congo red (C and D) staining. The endoneurium shows amorphous, eosinophilic hyaline deposits that are positive for Congo red.")
TTR gene sequencing was ordered, revealing the Val50Met mutation in heterozygosis.
Patient 3The patient was a 32-year-old woman from Portugal; her deceased father had been diagnosed with TTR-FAP due to the Val50Met mutation. Her father's 4 siblings (2 men and 2 women) and both paternal grandparents displayed the same disease. She attended our department in 2006 for a genetic study. The physical and neurological examinations and laboratory and neurophysiological studies performed at the time yielded normal results. The genetic study detected the same mutation in heterozygosis. The patient received genetic counselling and preimplantation genetic diagnosis, as she intended to have children. In July 2015, she attended a consultation at our department due to intense low back pain radiating to both lower limbs, together with burning and tingling sensations, allodynia, and oedema in the feet. A month earlier, she experienced alternating diarrhoea and constipation and marked abdominal distension. The patient reported instability and dizziness upon walking. She had no other associated symptoms, and presented no signs or symptoms of orthostatic hypotension. She was receiving treatment with venlafaxine, alprazolam, and mirtazapine due to reactive anxiety in response to the presymptomatic diagnosis of the disease. The neurological examination found normal touch, pain, and vibration sensation; deep tendon reflexes; and muscular balance. The echocardiogram revealed left atrial enlargement, with no additional alterations. A Holter ECG study detected infrequent ventricular extrasystoles. A bone scan revealed no abnormalities. Suspecting initial TTR-FAP symptom manifestation, we started treatment with tafamidis, plus duloxetine for pain control. As duloxetine was unsuccessful, we later prescribed eslicarbazepine; this was also ineffective, and substituted with tapentadol and diazepam. In February 2016, the patient displayed thermal hypoaesthesia in the feet and distal third of the legs; no other abnormalities were found. Given the persistence of intense, refractory neuropathic pain, a liver transplant was ordered in February 2016; the operation was performed 2 weeks ago.
Patient 4The patient was a 64-year-old former smoker from Gipuzkoa, with no relevant personal history. He attended the neurology department in March 2005 due to cramps and allodynia in the soles of the feet and difficulty climbing stairs, which had started 3 years earlier. His younger brother and deceased sister had both experienced similar symptoms and been diagnosed with TTR-FAP. Both siblings underwent liver transplantation. The neurological examination was normal except for reduced touch and vibration sensation in a “stocking” distribution, absent Achilles reflex, and patellar tendon hyporeflexia. A complete blood count detected no abnormalities. A neurophysiological study revealed axonal sensorimotor polyneuropathy. A genetic study identified the Val50Met mutation in heterozygosis. Holter ECG monitoring showed frequent supraventricular extrasystoles with multiple salvos of supraventricular tachycardia. No alterations were detected in the tilt-table test. We attempted to control neuropathic pain with pregabalin and amitriptyline. Symptoms continued to progress, with the patient developing motor deficits in the lower limbs, affecting gait. Despite a liver transplant performed in 2007, neuropathic pain continued to increase in intensity; duloxetine was added to the patient's treatment schedule. The patient developed constipation and oedema of the feet. The neurological symptoms remained largely stable, with distal weakness in the hands and feet and neuropathic pain; the patient was able to walk with a cane. Cardiac involvement progressed, and the patient died of heart failure 9 years after diagnosis.
DiscussionThe most common manifestation of TTR-FAP, described by Andrade in the Portuguese variant caused by the Val50Met mutation, is a sensorimotor polyneuropathy presenting with autonomic dysfunction, with onset occurring between the ages of 30 and 40 years.2–8,10 The condition initially affects the small myelinated (Aδ) and non-myelinated fibres responsible for transmitting pain and temperature signals.2,3,5 The greater the length of an axon, the greater its exposure to amyloid deposition and the resulting functional alteration.2,5 This explains the way the disease progresses, initially involving distal segments and eventually affecting more proximal areas.2,3,5,7 The natural history of the disease leads to death at 10-15 years after symptom onset.7,9,10
The 4 cases presented above demonstrate that TTR-FAP progression does not always follow this pattern. Symptom onset may occur late (in patients older than 50), and the disease may develop as purely motor or sensory polyneuropathy, with no autonomic dysfunction.2–5,10
This is the most frequent form of presentation in non-endemic areas. Another form is mononeuropathy progressing to mononeuritis multiplex and eventually a pattern of polyneuropathy.2–4,8,10 Neuropathic pain, which may appear at onset or later in disease progression, is a burning pain associated with allodynia, and exhibits a circadian rhythm, worsening in the evening.2,8,10 In some cases, such as the first patient's aunt, the mutation is non-penetrant.
TTR-FAP is a genetic but not necessarily a familial disease. Given the autosomal dominant inheritance pattern, family history of the condition assists in diagnosis; this was the case in 3 of the 4 patients presented.2–5,7 Family history of the disease is present in the majority of cases with onset at around the age of 30 years, whereas late-onset cases tend to be sporadic.11 The incomplete penetrance and the possibility of very late onset (with some individuals with the mutation dying before presentation of the disease) can give rise to the detection of apparently sporadic cases.2–5 There may also be cases where patients are unaware of their family history of the condition and do not report it.
TTR variants have been detected worldwide, especially in European countries, and display great genotypic heterogeneity. The genotype–phenotype correlation and the factors influencing phenotypic variability and age of onset are not fully understood.2,5 Amino acid substitution is not sufficient to explain the condition's variability in terms of penetrance, pathogenesis, and clinical progression.12 In order to better understand the genetic variability of TTR-FAP, haplotype characterisation should be performed for 6 single-nucleotide polymorphisms. In Portugal and Sweden, only haplotype I has been associated with the mutation. Haplotype III, the second most frequent variant, has been observed in individuals from Britain, France, Italy, and Japan.12 Soares et al.12 identified 10 new polymorphisms: 8 caused by single-base substitutions and 2 caused by insertions or deletions in dinucleotide repeat sequences. These authors hypothesise that age of onset may be modulated by a nearby locus linked to the interval defined by the microsatellites D18S457 and D18S456, associated with the 3-2 haplotype on the accompanying chromosome.12
A genetic anticipation phenomenon has been detected in some Portuguese, Swedish, and Japanese families with early-onset variants. This effect is more marked in cases where the mutation is transmitted by the mother.5 The literature includes reports of less severe phenotypes in cases with later onset and longer duration, in patients with such compound heterozygous mutations as Agr104His/Val50Met or Thr119Met/Val50Met.5,13 These additional mutations are believed to have a “protective effect” as they have been demonstrated to increase the stability of TTR tetramers.5,13
Diagnosis may be complex due to the disease's variability in terms of phenotype and age of onset. Conventional electrophysiological studies detect axonal neuropathy.3,5,7 Moderately reduced nerve conduction velocity is often misinterpreted as a form of chronic inflammatory demyelinating polyneuropathy, perhaps due to a desire to provide treatments for these patients. In the classic variants, electroneurography studies in early phases may display normal results, as the disease initially only affects small myelinated and non-myelinated fibres.2,5–7
Once polyneuropathy is confirmed, aetiology may be difficult to identify if we do not consider late-onset variants. The objective of tissue biopsy is to detect amyloid deposition. TTR amyloid deposits present greenish-yellow birefringence under polarised light following Congo red staining2,3,5,8; this is a pathognomonic feature of TTR-FAP. However, basing aetiological diagnosis on tissue biopsy has several limitations. Firstly, it is not clear which tissue should be examined, as we must select that which combines the best diagnostic yield with the lowest risk of undesired consequences.4,6–8 Given the relative aggressiveness of nerve biopsy, such other tissues as subcutaneous fat and salivary gland tissue have been proposed.1,4–8 Secondly, amyloid deposition is patchy; therefore, absence of amyloid deposition in the tissue sample tested does not rule out the condition.5,8 Finally, TTR amyloid deposition cannot be confirmed by the detection of amyloid deposits alone.6,8 Indeed, even if an immunohistochemical study determines that deposits are composed of TTR amyloid, it is not possible to differentiate between wild-type and mutant TTR.3,5,6,8
We therefore consider full TTR gene sequencing to be the diagnostic technique of choice1,3,5–8; due to the gene's short length and recent advances in genetic testing, this is an inexpensive, simple, quick technique with high sensitivity and specificity. Given that TTR-FAP can present with late onset and in the absence of known family history of the disease, especially in non-endemic areas, we believe that TTR sequencing should be included in diagnostic protocols for all polyneuropathies. If the disease is confirmed, family members at risk should also be tested, and the appropriate genetic counselling should be given.1,2,7,10
Comprehensive testing is also important, as this is a multisystem disease.7,8
Numerous treatments have been developed since 1990, with some still in the trial phase.14,15 The only treatments currently accepted are liver transplantation and tafamidis.
Liver transplantation is intended to prevent the formation of additional amyloid deposits (95% of mutant TTR is synthesised in the liver).2,8 The technique is intended to stabilise symptoms, but does not correct existing lesions. According to data from the Familial Amyloidotic Polyneuropathy World Transplant Registry, the survival rate following liver transplantation is 85% at 5 years, 67% at 10 years, and 55% at 20 years.8,16,17 The early-onset form of the Portuguese Val50Met variant responds best to treatment, with a 10-year survival rate of 74%.2,15 However, other mutations are associated with poorer outcomes,15 as are late-onset forms caused by the Val50Met mutation, which have a 10-year survival rate of 44%.10,13,16,18 Deposition of wild-type TTR may continue after liver transplantation, and can cause both neuropathy2,8,18 and cardiomyopathy.1,2 Cardiac amyloidosis progression appears to cease after liver transplantation in patients with early-onset TTR-FAP caused by the Val50Met mutation.1,19,20
Tafamidis stabilises TTR by inhibiting tetramer dissociation, slowing the progression of the disease.1,2,21,22 The drug performed better than placebo at 18 months in a double-blind randomised controlled trial including a sample of patients with early-stage TTR-FAP caused by the Val50Met mutation.1,2,8,23 Based on these results, the drug was approved by the Spanish Agency for Medicines and Medical Devices for the treatment of early-stage symptomatic TTR-FAP caused by the Val50Met mutation.1,8 It has also been studied in an open-label trial including 21 patients with other mutations; tetramers were observed to have stabilised after 6 weeks of treatment.24 There is no evidence of the drug's efficacy at later stages of the disease.
Besides these treatments, it is essential to manage multisystem complications.
ConclusionTTR-FAP is a complex, multisystem disease that is highly variable in terms of presentation and age of onset. Given its high sensitivity and specificity, full sequencing of the TTR gene should be included in diagnostic protocols for all patients with progressive polyneuropathies. Some treatments are known to be efficacious in treating the Portuguese variant of the disease, but their usefulness for other phenotypes is not known; future research should address this matter.
FundingThis study has received no funding of any kind.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Andrés N, Poza JJ, Massó JFM. Polineuropatía amiloidótica familiar I por mutación Val50Met (Val30Met) en el gen de la transtiretina. Neurología. 2018;33:583–589.
This study was awarded the prize for Best Oral Presentation at the 37th Meeting of the Neurology Society of the Basque Country, held at Hotel Monte Igueldo in San Sebastián on 26 and 27 February 2016.
recomendados

Neurología (English Edition) sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas