



The term focal cortical dysplasia (FCD) describes a particular migration disorder with a symptomatology mainly characterised by drug-resistant epileptic seizures, typical neuroradiological images, and histological characteristics, as well as a very positive response to surgical treatment in the majority of cases.
Material and methodsA total of 7 patients were studied, comprising 6 children with a mean age of 34.3 months and one 25-year-old male with very persistent focal seizures and MRI images that showed FCD.
ResultsThree of the patients (all girls) were operated on while very young, with extirpation of the FCD and the surrounding area; with the histopathology study showed agreement between the MRI images and the macroscopic study of the slices. The histology study showed findings typical of a Taylor-type FCD (poor differentiation between the cortical grey matter and the subcortical white matter, and balloon cells). Three years after the FCD extirpation, the same 3 patients remained seizure-free with no anti-epilepsy medication. Two others have seizure control with medication, another (the adult) is on the surgical waiting list, and the remaining patient refused the operation.
ConclusionTaylor-type FCD is associated with a high percentage of all drug-resistant focal seizures, and it needs to be identified and extirpated as soon as possible. Well planned and well-performed surgery that leaves no remains of dysplasia can cure the disease it in many cases.
El término displasia cortical focal (DCF) expresa una patología muy particular de trastorno de la migración que conlleva una sintomatología caracterizada principalmente por crisis epilépticas fármaco-resistentes, unas imágenes neurorradiológicas y unas características histológicas peculiares, así como una respuesta al tratamiento quirúrgico muy positiva en la mayoría de los casos.
Material y métodosSe estudia a 7 pacientes, 6 niños con edad promedio de 34,3 meses y un varón de 25 años con crisis focales muy rebeldes e imágenes de RM que mostraban DCF.
ResultadosTres de los pacientes (todas niñas) fueron operadas en edades muy tempranas, con extirpación de la DCF y la zona circundante, demostrando el estudio anatómico la concordancia de las imágenes de RM con las macroscópicas de los cortes anatómicos. El estudio histológico mostró los típicos hallazgos de la DCF tipo Taylor (mala delimitación entre sustancia gris cortical y la sustancia blanca subcortical, y «células balonadas»). Tres años después de la resección de la DCF los 3 pacientes estaban curados de las crisis y sin medicación antiepiléptica. Dos de los pacientes están controlados de las crisis con medicación, otro (el adulto) está en espera de decisión quirúrgica y el restante desechó la operación.
ConclusiónLa DCF tipo Taylor es una patología asociada a una buena parte de las crisis focales fármaco-resistentes, que debe tratarse de identificar y de extirpar lo antes posible ya que la cirugía, bien proyectada y realizada, sin dejar residuos displásicos, puede curarla en un alto porcentaje de casos.
The term ‘focal cortical dysplasia’ primarily refers to histological abnormalities found in the cortex of surgical specimens removed from the brains of subjects with drug-resistant epilepsy.1–3 The histological description of such lesions is a collection of large, strangely formed neurons with grotesque cells in some cases. These collections are located in deep areas of the cortex and the underlying white matter. Severity of FCD is related to its location, its gross morphology, and its histological characteristics. The mildest form of FCD is known as microdysgenesis, and some cases cannot be detected by neuroradiological imaging. The lesions are most commonly found in autopsies of epilepsy patients. They can now be found in epileptogenic zones in surgical specimens as well, thanks to the highly sensitive diagnostic imaging techniques that are currently available. The extension of FCD lesions varies; some may be found only in part of a fold, while others may affect an entire lobe. When they are found throughout an entire hemisphere or in parts of both hemispheres, the condition is called massive CD (MCD). Over the last few years, the terms ‘focal cortical dysplasia’,4,5 ‘cortical dysgenesis’,6 and the generic term ‘neuronal migration disorders’7,8 have become widely used. Some authors believe that the term FCD is the best one to designate the histopathological changes occurring in this disorder.9 Scientific literature search engines such as PubMed often include tubers, typical lesions of the cerebral or even cerebellar hemispheres that are concomitant with tuberous sclerosis complex (TSC),10 among forms of cortical/subcortical dysplasia. Although these syndromes can be differentiated clinically due to the conditions of the skin and other organs exhibited by TSC patients, MR imaging studies and histological characteristics for both FCD and TSC may be very similar in many cases.
The main clinical symptoms caused by FCD are epilepsy, focal neurological deficits, intellectual disabilities, delayed development of cognitive facets, or deterioration of those facets. For most focal-type cases, seizures may begin at any age, including during gestation, and continue throughout the subject's life, although they are more common in childhood. Seizures may be partial simple, partial complex, or generalised, depending on the FCD location and the patient's age. FCD is gaining increasing recognition as a cause of epilepsy. The lesion concomitant with FCD is rarely visible by computed tomography, at least when using older equipment. However, high-resolution magnetic resonance and some of its specific sequences11–20 often show cortical and subcortical lesions. Lesions are very frequently associated with hypoplasia of the hemisphere in which they are located, especially when they reach a certain size. Early diagnosis of these cortical malformations results in the decision to remove them surgically as soon as possible in order to prevent the short-term and long-term consequences associated with suffering a lengthy series of seizures.
In recent years, diagnostic imaging techniques have been perfected, and doctors are now able to identify dysgenic cortical or dysplastic epileptogenic tissue during surgical procedures. As a result, they can remove it more precisely and achieve better seizure control without increasing the size of the surgical lesion.20,21
Materials and methodsThe introduction of surgical treatment programmes, especially those for FCD patients, coincided with the policy of systematically performing 1.5T and 3T MRI studies on patients with focal epileptic seizures. Since that time, we have studied 7 patients with FCD and drug-resistant epilepsy at the Paediatric Neurology Department at Hospital Universitario La Paz in Madrid. Since about 1990, more than 150 cases of neuronal migration and cortical organisation disorders have been identified. The diseases causing these disorders were categorised mainly according to the characteristics displayed in the imaging studies.2,7,9,10,13,15–17 Here, we attempt to identify different entities using MRI studies with all of the sequences available to us, including T1, T2, contrast enhancement, inversion recovery (FLAIR), proton density, 3D MRI, diffusion-weighted MRI (especially axial), cortical surface reconstruction, and at times, MRI spectroscopy (this last technique was only used when it was necessary to obtain data that could distinguish between a malformation and a tumour). Functional imaging studies (mainly SPECT and PET) were also completed in some cases. Patients were selected based on presence of focal seizures that were resistant to specific antiepileptic drugs (administered in monotherapy, bitherapy, or even tritherapy in 1 case, during sufficiently long periods and at high enough doses to be able to determine whether or not the treatment provided effective seizure control); presence of focal epileptogenic activity whose location remained unchanged according to several EEG studies taken while doctors were attempting to control epileptic seizures; and presence of cortical dysplasia revealed by different neuroimaging studies. Regarding age, 6 of the 7 patients are children, and the seventh is a young adult who began suffering epileptic seizures in childhood. No cases of TSC were included.
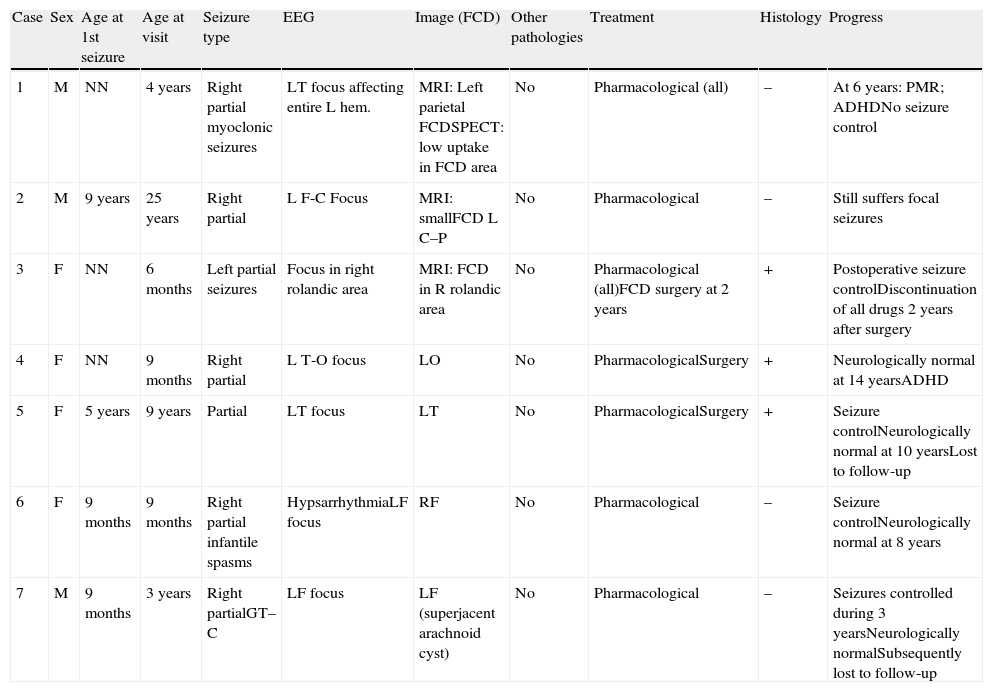
ResultsThis study focuses exclusively on 7 patients, comprising 6 children (2 males and 4 females) aged between 6 months and 9 years (mean age, 34.3 months) and an adult aged 25 years (Table 1). Imaging studies revealed FCD in all cases. Onset of seizures occurred during the first year of life in 5 paediatric patients, at the age of 5 years in the other child, and at the age of 9 years in the adult.
Focal cortical dysplasia (FCD).
| Case | Sex | Age at 1st seizure | Age at visit | Seizure type | EEG | Image (FCD) | Other pathologies | Treatment | Histology | Progress |
| 1 | M | NN | 4 years | Right partial myoclonic seizures | LT focus affecting entire L hem. | MRI: Left parietal FCDSPECT: low uptake in FCD area | No | Pharmacological (all) | – | At 6 years: PMR; ADHDNo seizure control |
| 2 | M | 9 years | 25 years | Right partial | L F-C Focus | MRI: smallFCD L C–P | No | Pharmacological | – | Still suffers focal seizures |
| 3 | F | NN | 6 months | Left partial seizures | Focus in right rolandic area | MRI: FCD in R rolandic area | No | Pharmacological (all)FCD surgery at 2 years | + | Postoperative seizure controlDiscontinuation of all drugs 2 years after surgery |
| 4 | F | NN | 9 months | Right partial | L T-O focus | LO | No | PharmacologicalSurgery | + | Neurologically normal at 14 yearsADHD |
| 5 | F | 5 years | 9 years | Partial | LT focus | LT | No | PharmacologicalSurgery | + | Seizure controlNeurologically normal at 10 yearsLost to follow-up |
| 6 | F | 9 months | 9 months | Right partial infantile spasms | HypsarrhythmiaLF focus | RF | No | Pharmacological | – | Seizure controlNeurologically normal at 8 years |
| 7 | M | 9 months | 3 years | Right partialGT–C | LF focus | LF (superjacent arachnoid cyst) | No | Pharmacological | – | Seizures controlled during 3 yearsNeurologically normalSubsequently lost to follow-up |
R: right; FCD: focal cortical dystrophy; Fr: frontal; L: left; F: female; O: occipital; P: parietal; C–P: central–parietal; NN: neonate; PMR: psychomotor retardation; T: temporal; GT–C: generalised tonic–clonic; ADHD: attention deficit hyperactivity disorder; M: male.
The clinical sign common to all cases was focal crises with different clinical expressions, the most common of which was myoclonic movement in a specific region of the body (lips, an upper or lower limb, etc.). In some cases, movements lasted only briefly; however, on rare occasions, they propagated to the entire side of the body without becoming generalised, lasting only a few seconds without the patient's losing consciousness and resolving without antiepileptic drugs. This occurred in our single adult patient (case 2). On other occasions, they evolved to become infantile spasms brought on by focal seizures. This occurred in case 6 in which simply administering an antiepileptic drug (Keppra) resolved the infantile spasms, corrected the hypsarrhythmia in the EEG, and controlled focal seizures. Eight years after treatment, seizures had not returned; a focus in the dysplastic area was still apparent, but the patient's neurological course was normal. Case 7 presented frontal focal dysplasia with a superjacent arachnoid cyst and focal seizures that were controlled at one point with antiepileptic drugs, but which became intractable in later years.
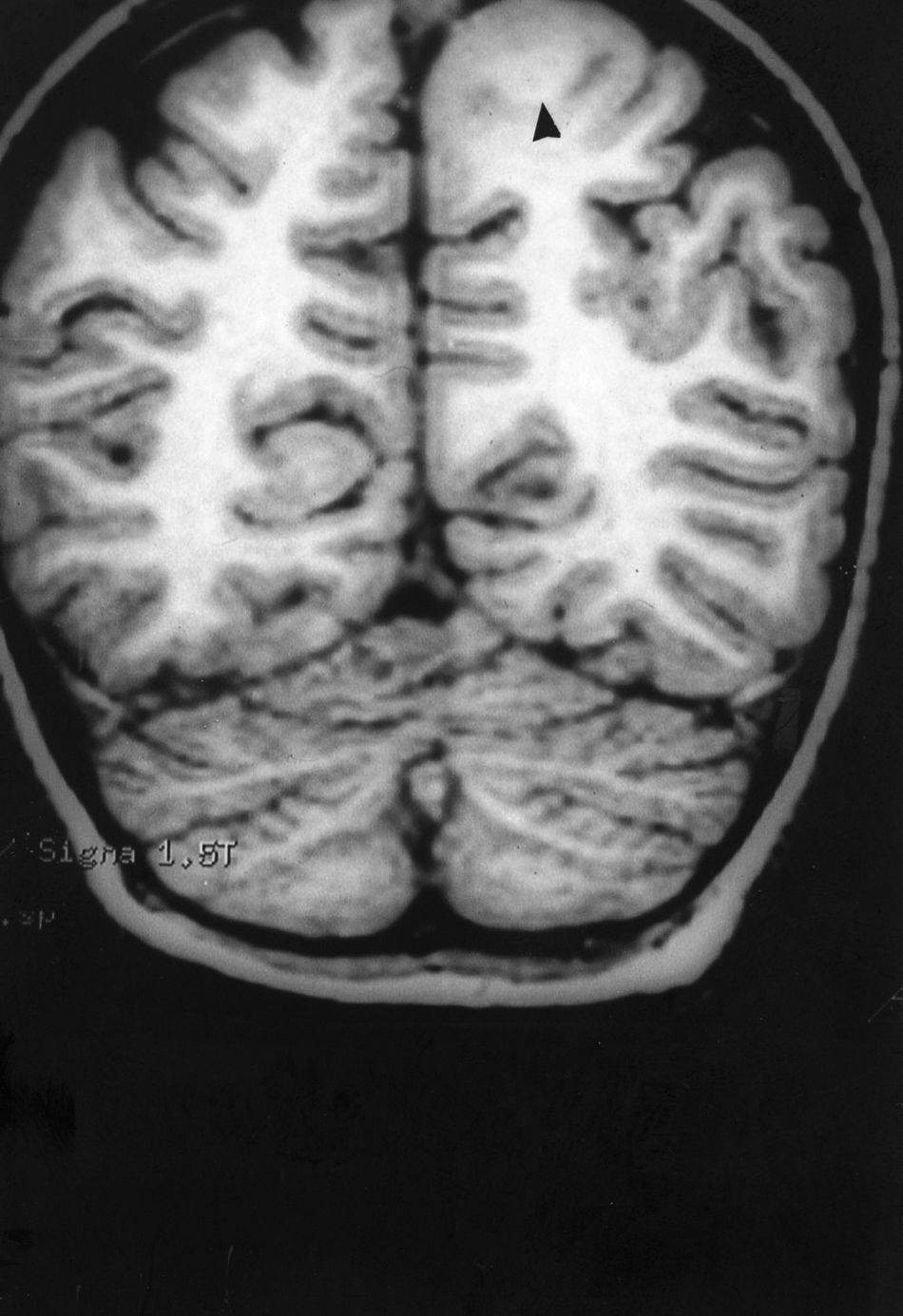
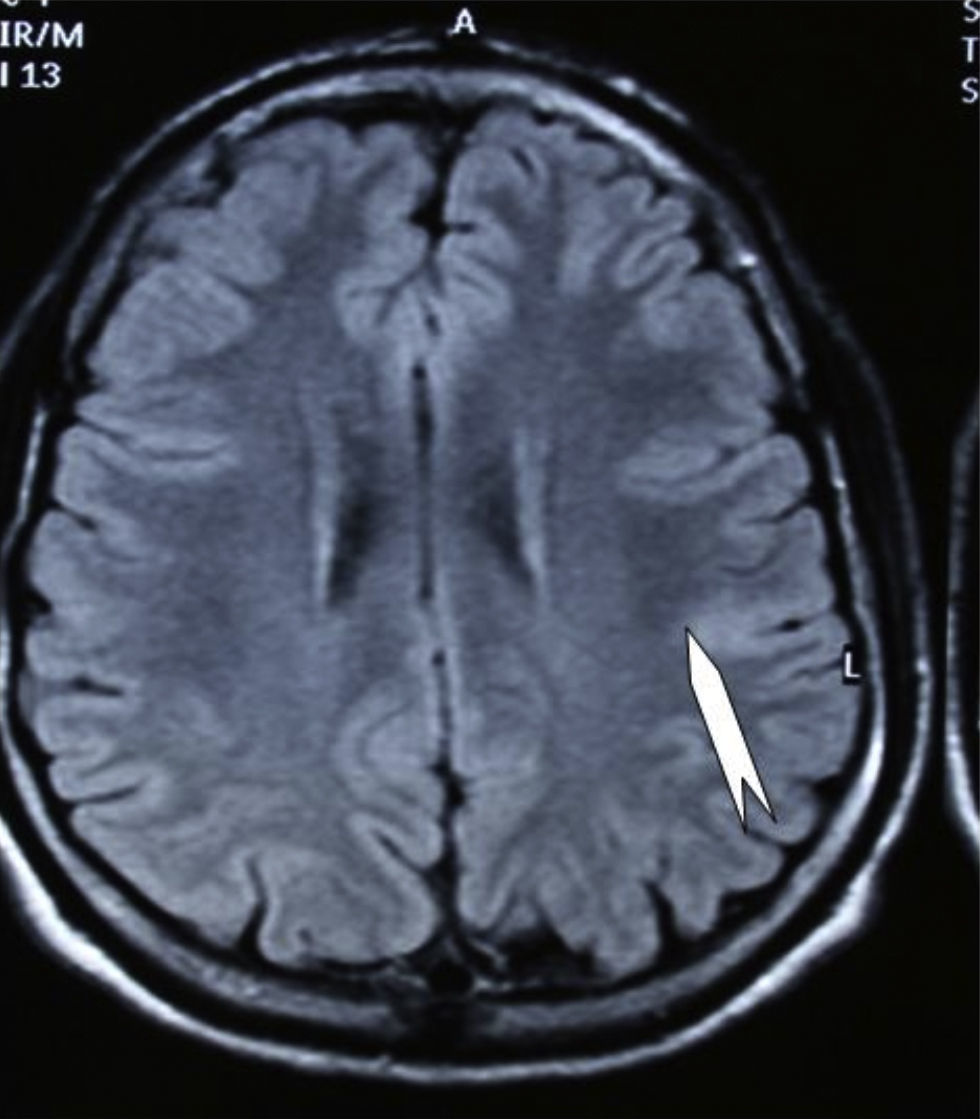
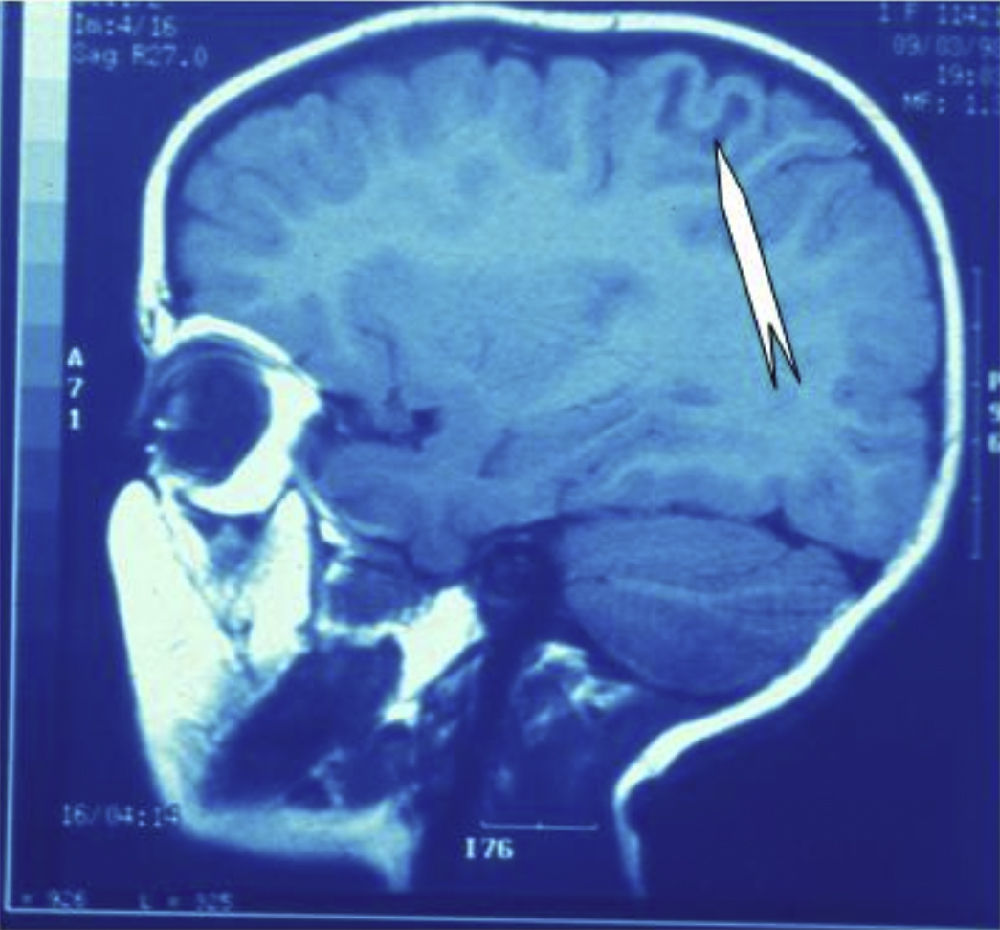
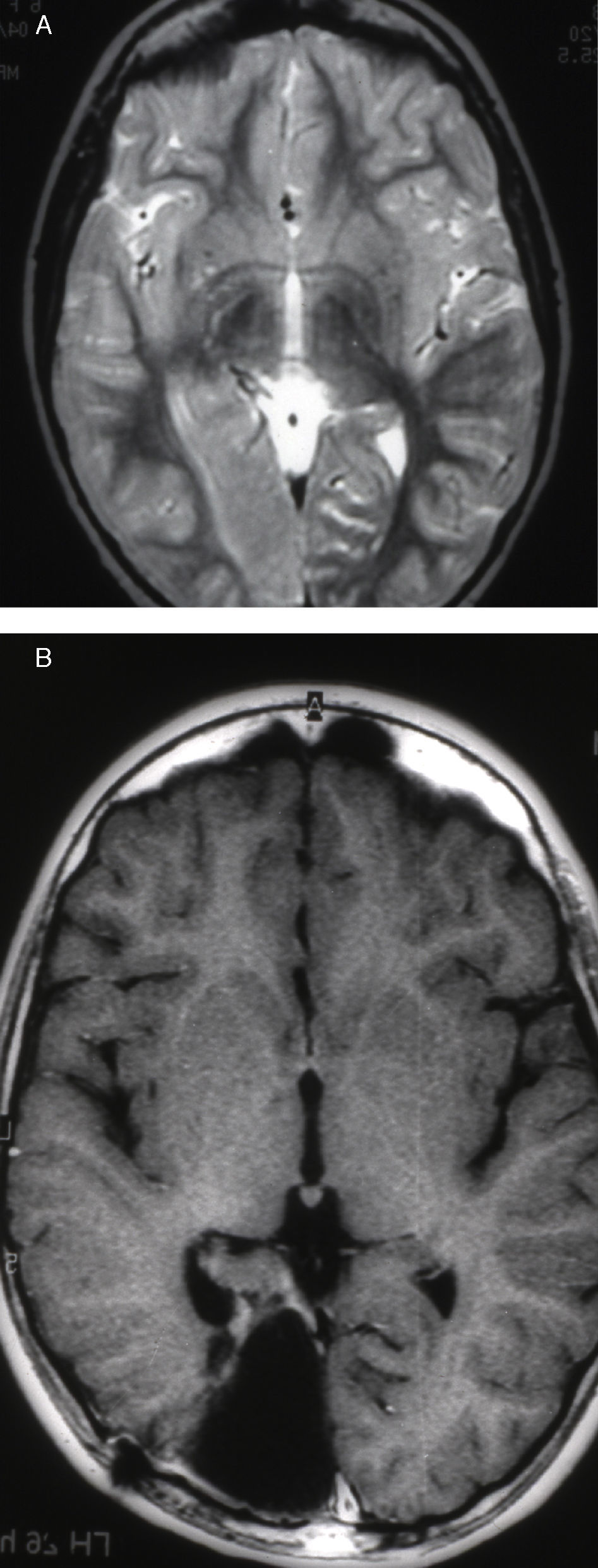
Seizures were more likely to appear during sleep in 2 patients, during waking hours in 3, and during either sleeping or waking hours in 2. Well-delimited and operable FCD was found in cases 3, 4, and 5, and the areas were extirpated completely. In cases 3 and 4, patients’ partial seizures with secondary generalisation disappeared completely, and these patients continued taking their previous antiepileptic medication during 2 years. At the end of those 2 years, doctors began to reduce medication, suspending it completely 3 years after the surgery. These patients have experienced no new seizures in the more than 10 years elapsed since that time. In case 5, seizure control was achieved by resecting the FCD; the patient remained seizure-free until he was lost to follow-up 3 years after his surgery. In case 1, displaying a medium-sized but very active FCD (Fig. 1) and a poor response to all types of antiepileptic drugs and numerous drug combinations, the family refused surgical treatment. When he was lost to follow-up at the age of 6, the child presented moderate psychomotor delays, restless personality, and uncontrolled seizures. Case 2, a 25-year-old male with a university degree, experienced 1 or 2 short rolandic seizures daily during sleep which did not cause either loss of consciousness or waking. Complete control could not be achieved using any of the drug combinations employed. He presented a small FCD in the rolandic area (Fig. 2) and has been scheduled for surgical treatment. Case 6 also has a small frontal FCD and his seizures have been controlled with antiepileptic drugs for the last 8 years. The patient is neurologically normal and his EEG shows foci. The MRI study shows well-delimited images of the FCD with good uptake of the paramagnetic contrast agents that show the size of the dysplasia precisely (Figs. 3 and 4).
. Note the thickening of the FCD, the loss of differentiation of cortical grey matter, the indistinct junction between the cortex and subcortical white matter, and signal changes inside the FCD.")
Case 1: Coronal 3D MRI slice. We observe a medium-sized FCD in the parasagittal region of the left posterior parietal lobe (arrow). Note the thickening of the FCD, the loss of differentiation of cortical grey matter, the indistinct junction between the cortex and subcortical white matter, and signal changes inside the FCD.
 in the left rolandic area.")
.")
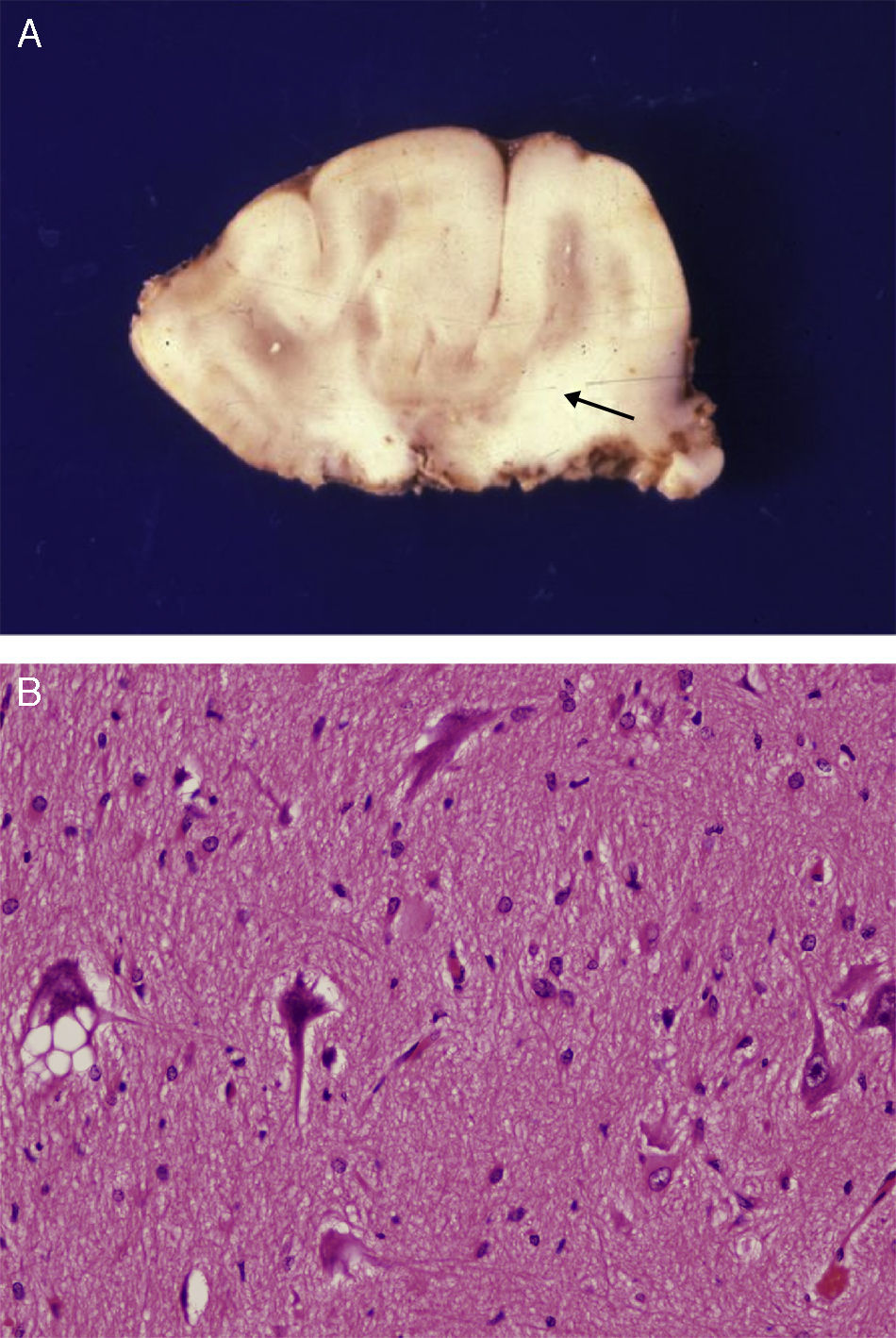
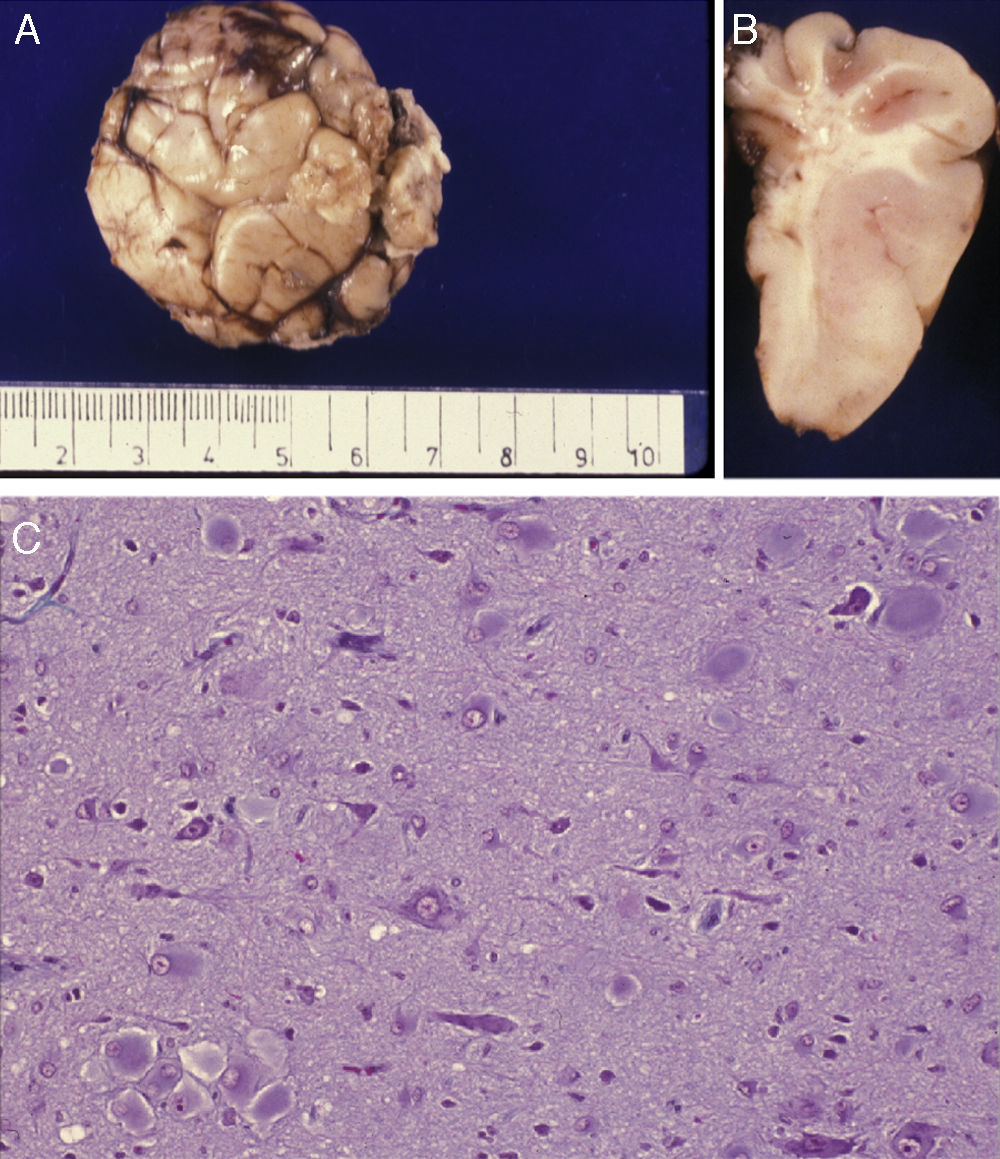
 Extracted neurosurgical specimen. The cortex displays variations in width and the white matter shows areas of subcortical demyelination tracing a U-shape, as was apparent on the MRI scan (arrow). (B) Histological study of the cerebral cortex showing structural changes, dysmorphic and poorly distributed neurons, and reactive gliosis. H&E stain 200×.")
Anatomical and histological study of the surgical specimen from the same patient. (A) Extracted neurosurgical specimen. The cortex displays variations in width and the white matter shows areas of subcortical demyelination tracing a U-shape, as was apparent on the MRI scan (arrow). (B) Histological study of the cerebral cortex showing structural changes, dysmorphic and poorly distributed neurons, and reactive gliosis. H&E stain 200×.
There was a high degree of correlation between neuroradiological images of the FCD and the anatomical appearance of the surgical specimen (Figs. 5 and 6). The MRI showed local cortical and subcortical signal changes which were generally small or medium-sized, with poor delimitation of cortical grey matter and subcortical white matter. Excised tissue specimens from the 3 cases treated surgically had the following dimensions: 5cm×3cm×2.8cm, weighing 20g (case 3); 5.5cm×5cm×2.5cm, weighing 33g; (case 4) and 6nm×3nm×2nm (case 5). Examination of some of these tissue samples reveals that at least 1 of the folds is larger and has a firmer consistency than the others. Multi-slice imaging of this larger fold shows an indistinct cortex/white matter junction caused by thickening of the cerebral cortex, which could be as much as 1cm thick. The demarcation between the cortex and the white matter is only blurred at certain points. The histological descriptions of the surgical specimens from each of the 3 patients are quite similar and a summary appears below. Their most remarkable feature is the presence of large atypical neurons grouped together in multiple foci and which disrupt the pattern of the normal laminar structure of the cortex. These neurons display morphological variations in their cytoplasm and frequently show signs of chromatolysis. Their distribution was also irregular. Other large cells with globoid nuclei and astroglial histogenesis were also present. Observation of the white matter showed both isolated heterotopic cells and cell clusters representing the 2 types of cell described above. They were also present in the molecular layer of the cortex, where clear marginal gliosis could be observed. White matter adjacent to the cortical changes described above explains the abnormally thick folds that could be observed macroscopically.
 and after (B) surgical resection.")
 External appearance of the surgical sample shows severely deformed folds and sulci. (B) Lateral slice of the surgical sample shows irregular cortical thickness with highly myelinated white matter. (C) Histological preparation in which we observe isolated or grouped balloon cells in the cerebral cortex. Masson")
Anatomical and histological study of the surgical sample from the same patient shown in Fig. 5. (A) External appearance of the surgical sample shows severely deformed folds and sulci. (B) Lateral slice of the surgical sample shows irregular cortical thickness with highly myelinated white matter. (C) Histological preparation in which we observe isolated or grouped balloon cells in the cerebral cortex. Masson's trichrome stain 200×.
Definitive control over seizures was achieved by surgery in all 3 cases treated surgically (antiepileptic drugs were provided for 2 years, after which they were discontinued).
DiscussionCortical malformations, which frequently cause drug-resistant epilepsy, have been described under a number of names. The generic term ‘cortical dysplasia’ is frequently used to describe malformations of cortical development that include heterotopias, polymicrogyria, and FCD. The best-known form of FCD is that described by Taylor,1 and it is characterised by the presence of balloon cells. This type of cortical anomaly is therefore known as Taylor's FCD or balloon-cell subtype. Different types and subtypes of FCD have been described based on histological changes in the cortex of patients who have undergone surgical treatment for intractable epilepsy. Each one can be distinguished from the others due to histological particularities corresponding to either FCD type II or Taylor's FCD.21 Children with Taylor's FCD experience early-onset intractable epilepsy with mild neurological comorbidities. The condition responds well to antiepileptic drugs, but patients with the FCD subtype that is not accompanied by balloon cells display a more severe phenotype.22 Categories based on imaging, which several authors described a few years ago under the less precise name ‘malformations of cortical development’,2,23–25 are more generalised and include numerous types of cortical abnormalities; under these criteria, FCD is grouped with another type of malformations that are similar to one another.26
The 2 types of FCD, Taylor's FCD and non-Taylor's FCD, can normally be distinguished from one another by MR imaging. Characteristics suggesting Taylor's FCD include focal cortical thickening and indistinct grey/white matter junction that frequently tapers towards the ventricle.17 The lesion is generally extratemporal in Taylor's FCD and temporal in non-Taylor's FCD. The latter is frequently associated with hippocampal sclerosis.17 Identifying some types of FCD using low-resolution MRI was not easy, and small lesions sometimes went undiagnosed a few years ago. This rarely occurs now, as the fluid attenuated inversion recovery pulse sequence (FLAIR) detects most such lesions. However, some small lesions may still go undetected, and very large lesions may have indistinct edges. Because of this, in cases of suspected FCD, we recommend using as many MRI sequences as possible, especially 3D-FLAIR, proton-density weighted sequence (PD), and high resolution T2-weighted sequence for transverse slices.15 Using these techniques and a 3T MRI machine (if available)13, we are able to diagnose FCD more accurately and identify its type and size, enabling us to plan safer surgical procedures. As imaging techniques evolve, there are fewer cases of focal epilepsy of unknown aetiology and more cases of successful surgical treatment among patients with drug-resistant seizures. Since the introduction of new imaging techniques, we have learned that between 20% and 25% of patients with focal epilepsy have FCD27–29 and 76% of them experience drug-resistant seizures.30 The 2 types of FCD can now be distinguished from one another either by their histological characteristics (presence or absence of balloon cells) or by their innate, adapted immune characteristics.31 While these findings are not necessary for a diagnosis of FCD, they do allow us to categorise types of cells after diagnosis.21 Not all patients with focal seizures caused by FCD are candidates for surgery; in some cases the seizures can be controlled with drugs, at least for a few years. In others, including a patient in our series, critical external manifestations are mild. However, surgery is the option offering the best outlook for most FCD patients, especially those with intractable seizures.
Prior to the arrival of high-definition diagnostic methods, including a number of functional imaging techniques in the area of nuclear medicine, a lengthy list of MRI sequences, and the combination of different methods, cortical dysplasia was listed in large series among the rarer types of malformations of cortical development.32 Nevertheless, imaging and histopathological studies with better technical equipment, which have recently been carried out in patients with drug-resistant epilepsy,20–39 have increased the possibility of detecting both very small lesions and large lesions with indistinct areas. It has also become easier to distinguish between arrays of FCD-type anomalies and those pertaining to mesial temporal sclerosis or benign tumours, even when 2 of these entities may be present in the same patient. Functional images must be obtained prior to surgery if we are to generate a cortical map in order to prevent post-operative neurological deficits.39 This is almost always successful according to the results of our series. FCD's ability to provoke seizures is related to excitatory and inhibitory neurotransmission abnormalities, according to immunological and histochemical findings from histological tissue specimens from patients suffering from various types of seizures.40,41
The incidence of FCD in series of patients who undergo surgery for epilepsy ranges between 12% and 40%.28,42–44 Seizure control is achieved in 63% to 80% of all cases.33,34,45,46 In the case of the rolandic variant of FCD, which requires increased precautions to be taken during surgery so as not to leave lesions that could result in motor or sensory sequelae,47 59% of such cases are seizure-free after surgery.
One factor predicting favourable prognosis is total resection of the FCD according to the map of the lesion obtained by neuroimaging and electro-anatomical mapping techniques. The factor predicting the poorest outcome and seizure recurrence is incomplete resection of the lesion. For all varieties and locations of FCD, exacerbation of seizures can be observed in the immediate postoperative period, predominantly in the rolandic area. This is due to the increased difficulty of completely extirpating lesions at this location. Status epilepticus immediately following surgical resection of the lesion has been observed in 20% to 37% of cases with balloon-cell subtype FCD2,47,48 at any location. Some suggest that this condition could arise due to balloon cells in the FCD lesion playing an inhibitory role. When they are resected, it could trigger a release of excitatory factors contained in the tissue surrounding any remaining epileptogenic cortex and cause seizure self-limiting mechanisms to fail.47 We have also observed onset of status epilepticus immediately following the extraction of epileptogenic tubers in 2 cases of tuberous sclerosis complex (not included in this study). This is logical when we consider that the histopathological structure of cortical tubers may be indistinguishable from that observed in some forms of FCD; both diseases show large, strangely formed neurons, atypical astrocytes, and subpial fibrillary gliosis.49
The aetiology of type 1 FCD remains unknown. Some authors believe that it arises due to exogenous lesions, such as those left by hypoxia, infections during gestation, or perinatal trauma,50–52 but this theory has not been demonstrated conclusively.
Some types of neurological sequelae are common during the postoperative period. They include dysarthria, hemiparesis, monoparesis, specific intellectual deficiencies, and behaviour peculiar to attention deficit hyperactivity disorder (ADHD), as we have observed in certain cases in this series.
ConclusionWe selected a group of 7 patients with onset of focal seizures during childhood, very active foci in the EEG that did not change over time, and an image of cortical dysplasia from an imaging study employing multiple MRI techniques. Full surgical resection of the dysplasia resulted in seizure control and discontinuation of antiepileptic drugs in the 3 cases in which that technique was employed. Histological studies of the excised tissue samples revealed findings compatible with Taylor-type FCD. In 2 cases, seizure control was achieved after several attempts with different drug regimens. One patient with severely intractable seizures refused surgical treatment; another, now an adult in good neurological condition but with poor seizure control, has been scheduled for surgery. Drug-resistant seizures and signs of FCD in neuroimaging studies are crucial indicators of surgical treatment.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Pascual-Castroviejo I, et al. Displasia cortical focal. Correlaciones clínico-radiológicaspatológicas. Neurología. 2012;27:472–80.
