




Las ataxias hereditarias (AH) y paraparesias espásticas hereditarias son enfermedades raras, poco frecuentes en las consultas del neurólogo general. Proponemos una guía práctica y breve de diagnóstico y manejo de estos pacientes, así como un procedimiento para la evaluación integrada del grado de su discapacidad. Se describen por apartados los conceptos y definiciones de AH y AH-plus y paraparesias espásticas hereditarias pura y complicada, la valoración clínica de los pacientes con las principales pruebas complementarias a realizar, las escalas clínicas necesarias para poder graduar la condición física y psíquica de los pacientes, y se resumen los tratamientos disponibles. Esta guía pretende facilitar la asistencia clínica diaria por parte del neurólogo y unificar los criterios médicos y la metodología de evaluación de la discapacidad de los pacientes con AH y paraparesias espásticas hereditarias.
Hereditary ataxia (HA) and hereditary spastic paraplegia are rare diseases; as such, they are rarely managed in general neurology consultations. We present a set of brief, practical recommendations for the diagnosis and management of these patients, as well as a standardised procedure for comprehensive evaluation of disability. We provide definitions for HA and “HA plus,” and “pure” and “complicated” hereditary spastic paraplegia; describe the clinical assessment of these patients, indicating the main complementary tests and clinical scales for physical and psychological assessment of the patients; and summarise the available treatments. These recommendations are intended to facilitate daily neurological practice and to unify clinical criteria and disability assessment protocols for patients with HA and hereditary spastic paraplegia.
Las enfermedades raras se definen como aquellas que afectan a un número limitado de personas en el total de una población definida. En España y en Europa se consideran como tales aquellas que afectan a menos de 1 por cada 2.000 habitantes (prevalencia general del 6-8% de pacientes a nivel mundial).
En la actualidad hay más de 7.000 enfermedades raras descritas, y una gran parte afectan al sistema nervioso. Se estima que hasta el 65% de estas patologías conllevan un grado de discapacidad física o psíquica que limitan la autonomía del paciente.
Las ataxias hereditarias (AH) y las paraparesias espásticas hereditarias (PEH) son síndromes neurodegenerativos de base genética altamente heterogénea. La prevalencia es desconocida, pero se estima a nivel mundial en unos 3-20 enfermos por cada 100.000 habitantes para las AH, y 1,2-9,6 por cada 100.000 habitantes en el caso de las PEH. Pueden afectar a cualquier sexo y edad, predominando, en su conjunto, en el adulto joven.
Recientemente en España se ha publicado un mapa epidemiológico con las cifras de prevalencia estimada de las AH y PEH entre los años 2018 y 20191. La prevalencia estimada de AH en nuestro país es de 5,48 casos/100.000 habitantes, y la de PEH es de 2,24 casos/100.000 habitantes. La AH dominante más frecuente es la SCA3, seguida de la SCA2. Las AH recesivas más frecuentes son la ataxia de Friedreich, y el Niemann-Pick C. La PEH dominante más frecuente es la SPG4, seguida de la SPG17, y la PEH recesiva más frecuente es la SPG7, seguida de la SPG11. En el 47,6% no se ha conseguido un diagnóstico genético.
La nueva ley de discapacidad recientemente publicada (Ley 8/2021 de 2 de junio, BOE 3 de junio), recoge una serie de modificaciones para el apoyo a las personas con discapacidad2. Las AH y PEH son enfermedades raras, poco frecuentes en la consulta del neurólogo general o en la consulta de otros médicos que pueden valorar la discapacidad de estos pacientes (Atención Primaria, Medicina Interna, Medicina Física y Rehabilitación, Medicina del trabajo, Salud laboral, peritos médicos, etc.)3,4.
El objetivo de este documento es que pueda ser consultado por neurólogos y médicos relacionados con la valoración de los pacientes con AH y PEH, para facilitar el diagnóstico y la gradación de discapacidad de una manera adecuada y, asimismo, facilitar el acceso del paciente a los recursos sociales y legales acordes a su situación.
MétodosSe ha realizado una búsqueda de la bibliografía en base a la evidencia científica disponible en bases de datos como Pubmed, Cochrane Library Plus, Clinical Key, MESH, Medline, desde los años 80 hasta 2021. También, se han valorado las guías de práctica clínica y documentos de consenso más recientes.
Se ha desarrollado una breve guía orientada a la valoración clínica y diagnóstica, y un método que permite graduar la condición física y psíquica del paciente. Para esto, se han definido los conceptos de las AH y PEH, y se han seleccionado las escalas validadas más empleadas en la práctica clínica. A su vez, se han desarrollado unas tablas y diagramas de flujo eminentemente prácticos para facilitar el diagnóstico y la evaluación integral de la discapacidad del paciente, objetivo principal de este trabajo.
Hay múltiples clasificaciones de estas enfermedades según su patrón genético, tipo de instauración, edad de inicio, múltiples estudios complementarios, etc., que ayudan a seleccionar los estudios genéticos más adecuados. Así mismo, hay varias escalas validadas dirigidas a completar la evaluación física y psíquica de estos pacientes. Toda esa información excede el objetivo de esta guía breve y práctica para la consulta habitual, pero se puede consultar en la web de la Sociedad Española de Neurología: «Guía de evaluación diagnóstica y de discapacidad en pacientes con ataxias y paraparesias espásticas hereditarias» (https://www.sen.es/profesionales/guias-y-protocolos).
ResultadosDefinicionesAtaxia: trastorno de la coordinación por disfunción del cerebelo y sus conexiones.
Ataxia-plus: implica que, además de los síntomas de ataxia, se añaden otras manifestaciones clínicas por afectación de otras estructuras neurológicas y/o sistémicas (atrofia óptica, degeneración retiniana, trastornos oculomotores, epilepsia, mioclonus, signos piramidales, signos de polineuropatía, afectación multisistémica, etc.).
Paraparesia espástica hereditaria: grupo heterogéneo de enfermedades genéticas que cursan con espasticidad bilateral de las extremidades inferiores y debilidad, que provocan una dificultad para la marcha (clínica piramidal±disfunción esfinteriana±hipopalestesia distal de miembros inferiores).
Paraparesia-plus (complicada): implica que, además de la paraparesia espástica, se añaden otras manifestaciones clínicas como ataxia, epilepsia mioclónica, crisis, signos extrapiramidales, trastornos oculomotores, etc.
AnamnesisEn todos los pacientes con sospecha de ataxia, se debe realizar una anamnesis completa que debe incluir: historia familiar y existencia de consanguinidad; embarazo, parto y desarrollo psicomotor; trastornos de la marcha, del equilibrio o inestabilidad, alteración del habla (disartria), trastornos oculomotores como diplopia, oscilopsia y rigidez de las extremidades, o tropiezos5–7.
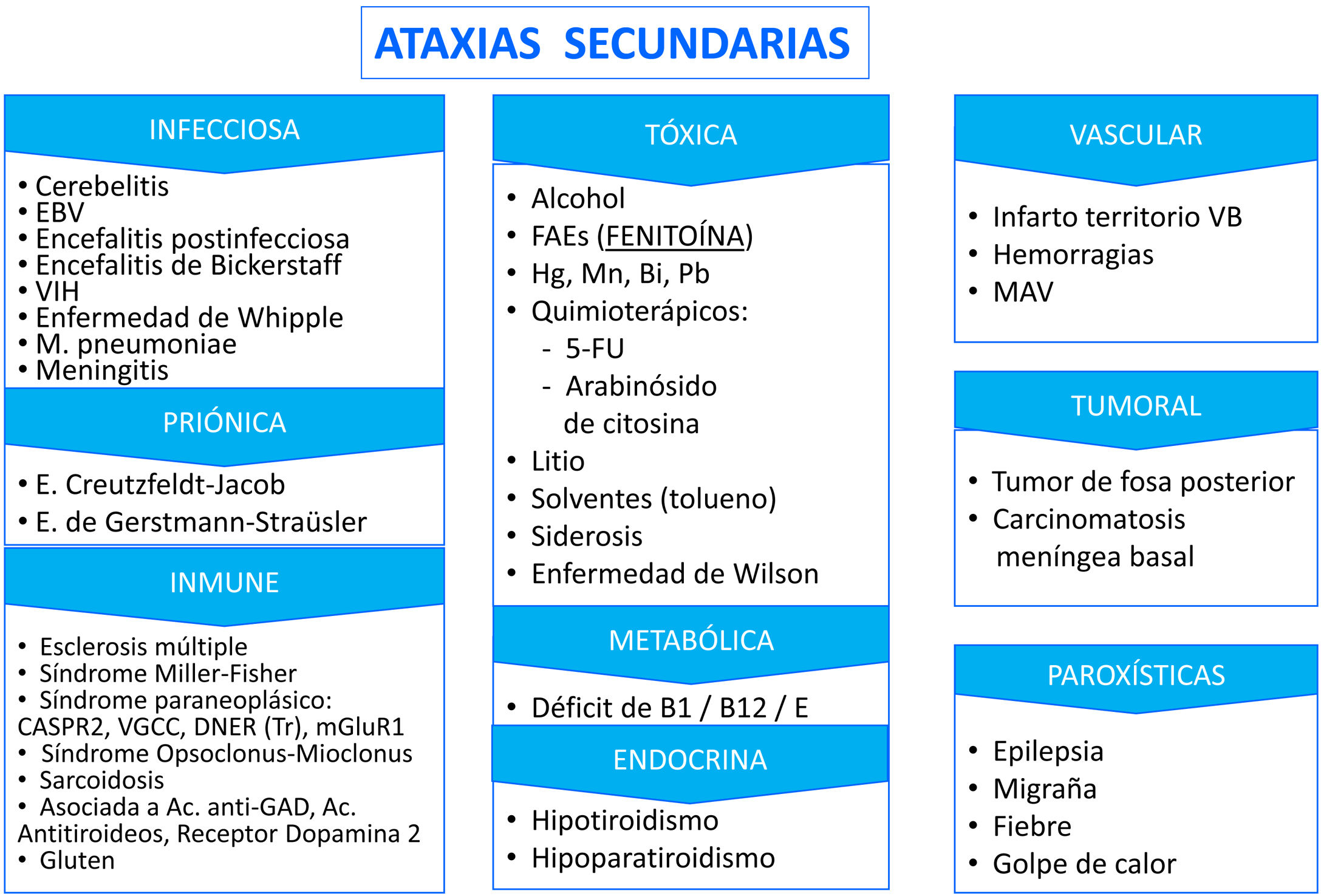
También debemos especificar la instauración (aguda, subaguda, o crónica) y evolución (lenta progresiva, rápidamente progresiva, episódica, estable), así como factores desencadenantes y agravantes, para poder diferenciar las ataxias adquiridas (tablas 1 y 2). Otras manifestaciones a valorar son: urgencia miccional-incontinencia urinaria, parestesias, espasmos musculares y dolor en miembros inferiores, y manifestaciones psiquiátricas y sistémicas. Es importante considerar los antecedentes de consumo de alcohol, drogas y fármacos.
Clasificación de las ataxias primarias Klogether 2005
| Ataxias hereditarias | Ataxias no hereditarias |
|---|---|
| Autosómicas dominantes (SCA) | Atrofia multisistémica tipo cerebeloso |
| Autosómicas recesivas (ARCA) | Atrofia cerebelosa tardía idiopática y esporádica |
| Ligadas al cromosoma X | Ataxias sintomáticas (degeneración cerebelosa alcohólica, tóxicos, paraneoplásica, déficits vitamínicos o trastornos metabólicos adquiridos, encefalitis cerebelosa inmunes |
| Episódicas |

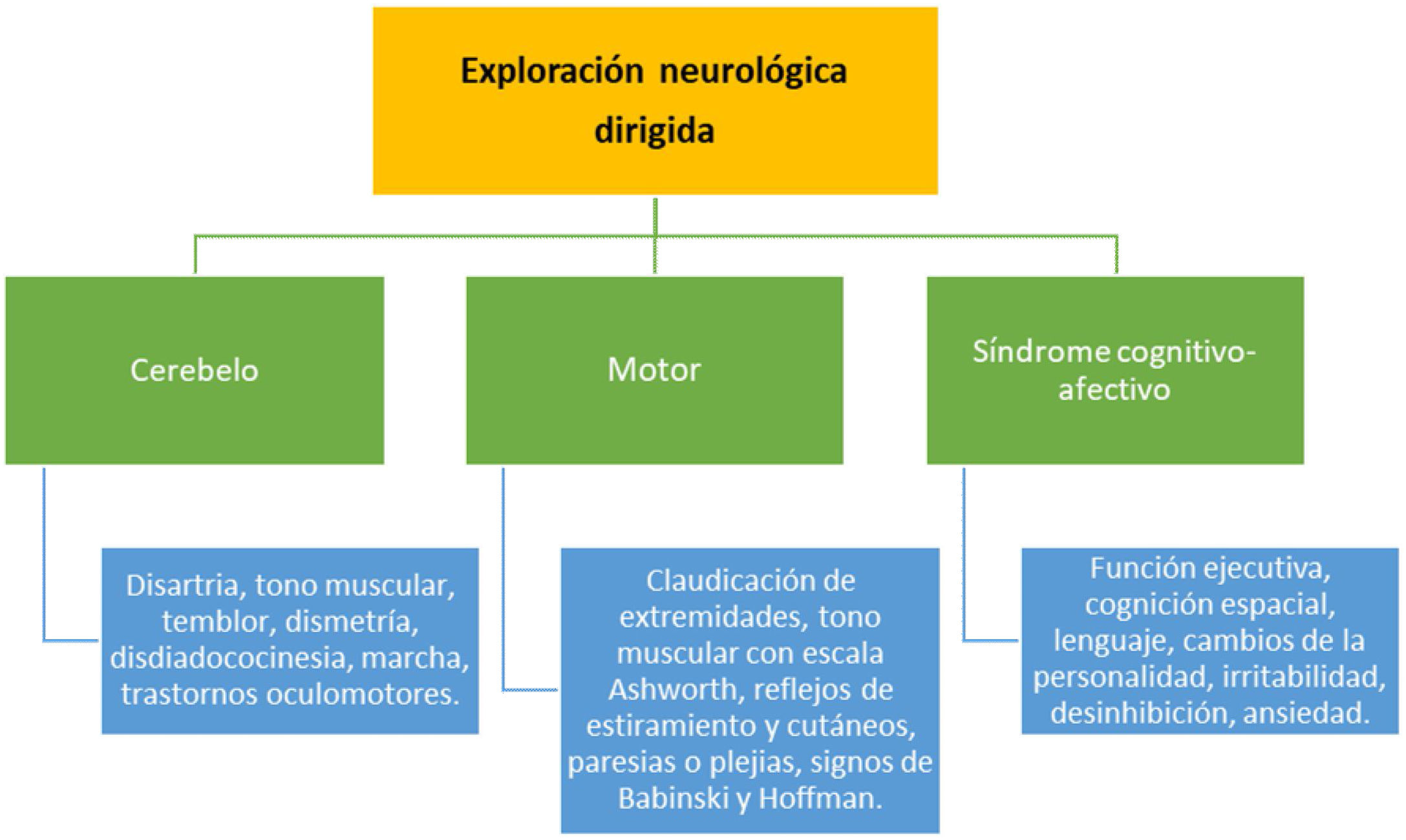
Exploración específica de cerebelo, sistema motor, y valoración cognitivo-afectiva (fig. 1). Todos estos puntos detallados deberían constar específicamente en la historia clínica de los pacientes.
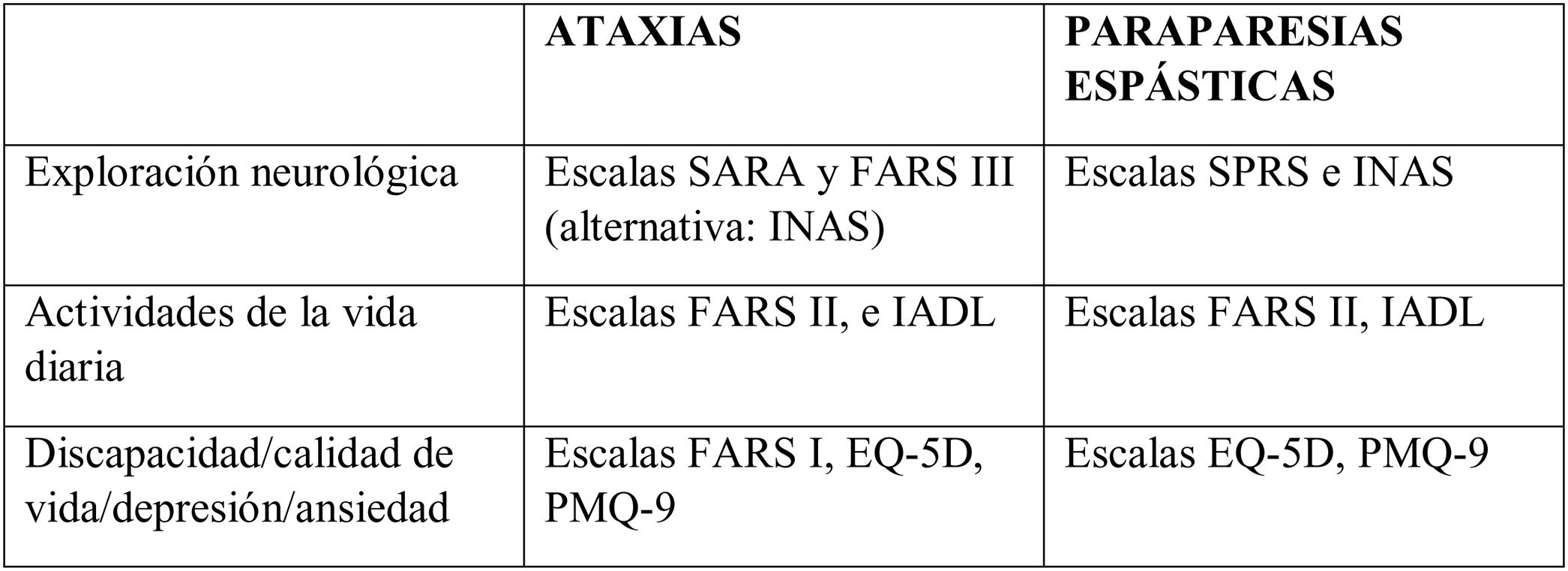
Escalas estándar para valorar la discapacidad
Las escalas más habituales y más empleadas, tanto en la práctica asistencial como en ensayos clínicos, son las siguientes7–20:
- -
Scale for the Assessment and Rating of Ataxia (SARA).
- -
Friedreich Ataxia Rating Scale (FARS) Functional Stage.
- -
Fenotipado sistemático utilizando el Inventory of Non-Ataxia Signs (INAS) con modificaciones particulares (p. ej., bradicinesia, ptosis o la prueba de impulso cefálico HIT).
- -
Spastic Paraplegia Rating Scale (SPRS).
- -
Activities of Daily Living (ADL).
- -
Cuestionarios sobre el estado de salud y la depresión (EQ-5D/EQ5D-Y y PHQ-9).
- -
Índice de gravedad de la enfermedad ARSACS: medida de resultado específica de la enfermedad.
Todas son laboriosas y requieren un tiempo de realización mayor de 15 min. Las más extendidas en la práctica asistencial son la SARA7 y la SPRS17 y son las que al menos deberían figurar en la historia clínica.
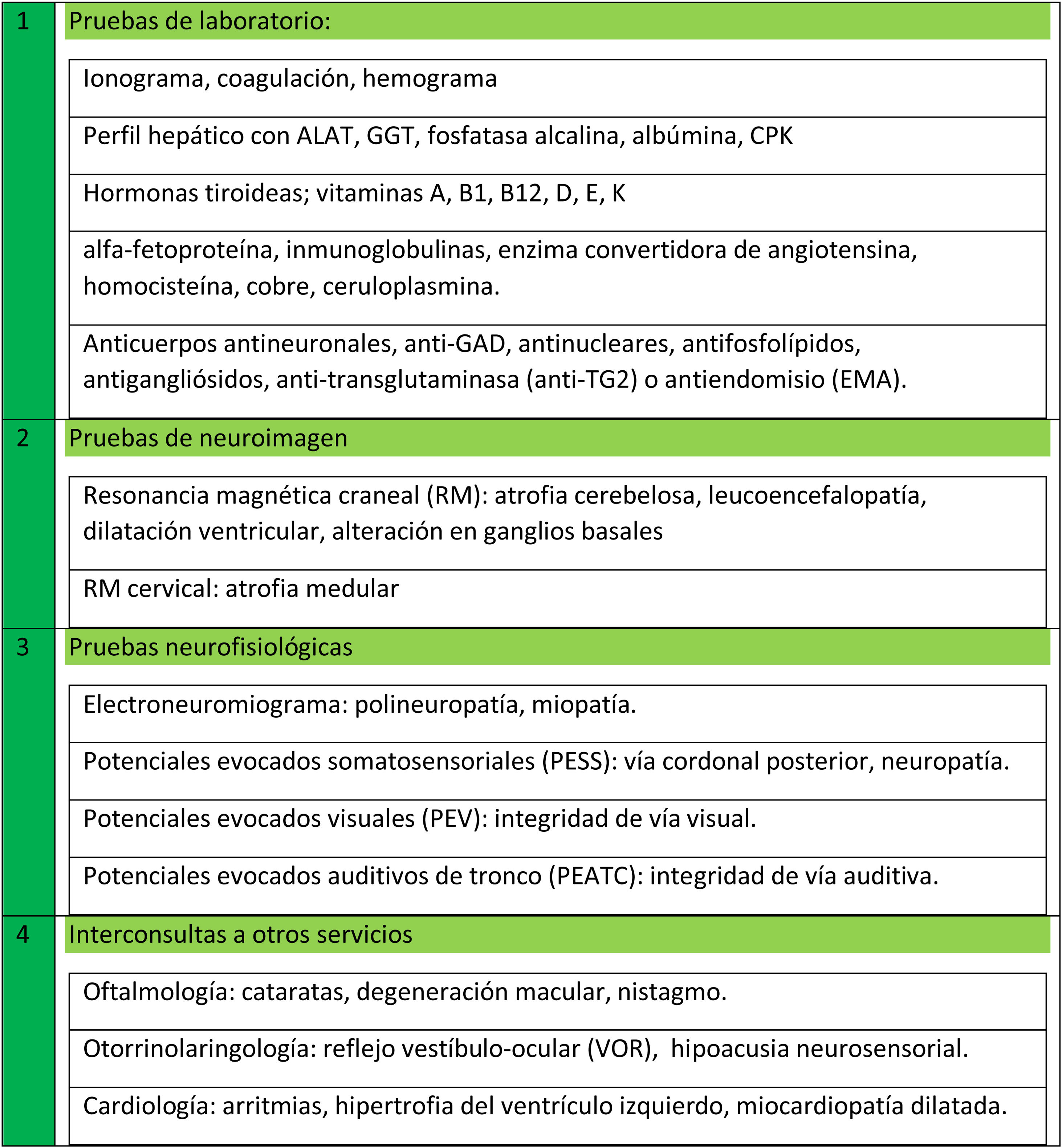
Pruebas complementariasDentro de los exámenes complementarios comunes en estos pacientes, es recomendable que dispongan de un estudio mínimo básico de pruebas, realizables en la mayoría de los centros sanitarios ambulatorios/hospitalarios como: resonancia craneal y cervical, electroneuromiograma, potenciales evocados (visuales, somatosensoriales y/o auditivos), valoración oftalmológica, cardiológica y otorrinolaringológica, y pruebas de laboratorio (fig. 2)5,20-23.

Los estudios metabólicos y hormonales pueden ser muy amplios, pero en general, las pruebas de laboratorio básicas que se deben incluir en todos los casos son:
- -
Ionograma, CPK, albúmina, CBC con frotis de sangre y búsqueda de acantocitos, ALAT, GGT, fosfatasa alcalina.
- -
TSH, T3+T4.
- -
Vitaminas A, B1, B12, D, E y K, alfa-fetoproteína, inmunoglobulinas, enzima convertidora de angiotensina, homocisteína, cobre, ceruloplasmina, albúmina.
- -
Anticuerpos antineuronales, anti-GAD, antinucleares, antifosfolípidos, antigangliósidos, antitransglutaminasa (anti-TG2) o antiendomisio (EMA).
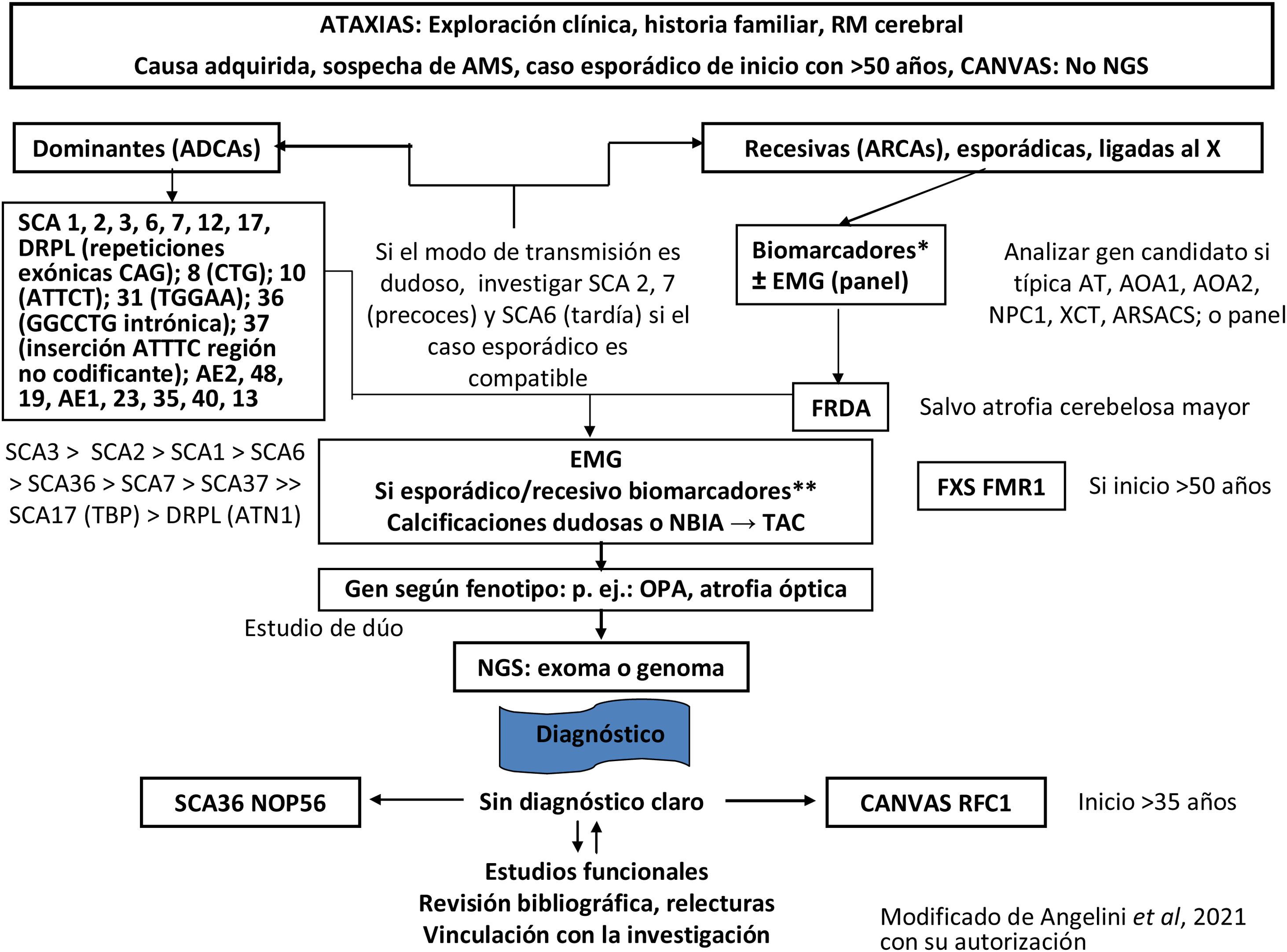
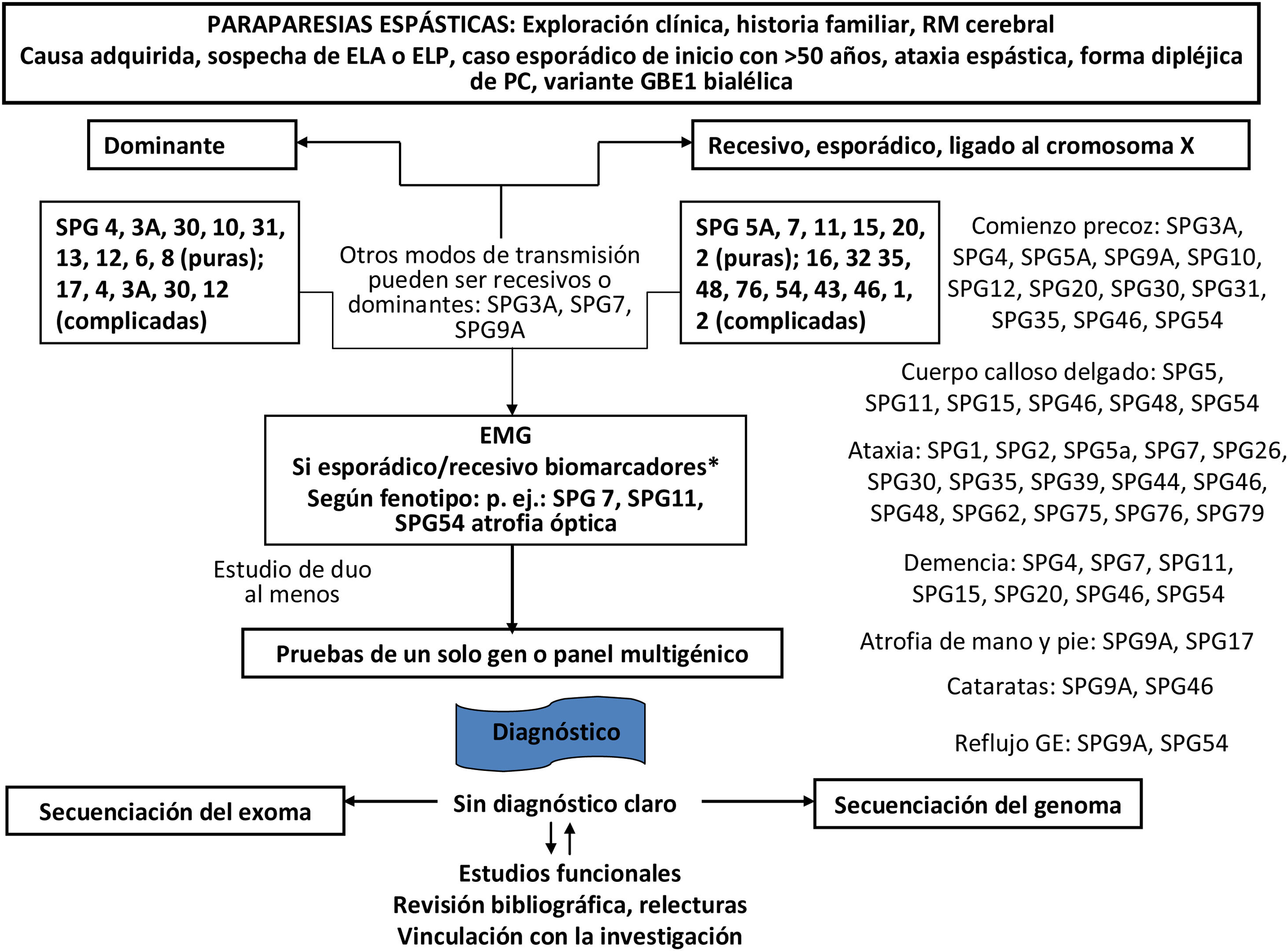
Finalmente, se intentará en la medida de lo posible filiar el diagnóstico genético molecular del tipo de AH o PEH. Tanto las AH como las PEH tienen formas hereditarias autosómico dominantes, recesivas, ligadas a X, y mitocondriales. El estudio genético se orientará dependiendo de la sospecha diagnóstica, pudiendo variar según la disponibilidad de técnicas y recursos en cada centro, y en las comunidades autónomas (figs. 3-5)5,20–22,24,25. Aun así, y a pesar de llevar a cabo un riguroso proceso diagnóstico, hasta el 40-50% de los pacientes no recibirán un diagnóstico definitivo1.
; NPC1: Niemann-Pick tipo C1; OPA: atrofia óptica; RFC1: subunidad C del factor de replicación (CANVAS); RMF1: retraso mental por cromosoma X frágil 1 (FXS); SCA: ataxia espinocerebelosa; TBP: proteína de unión a caja TATA (SCA17); TC: tomografía computarizada; XCT: cerebrotendinosis xantomatosa. * Biomarcadores 1: vitamina E, cobre y ceruloplasmina, alfa-fetoproteína, albúmina, hexosaminidasas, ácido fitánico, pristánico, ácidos grasos de cadena muy larga, homocisteína, colesterol, colestanol, Lyso SM509, creatina fosfoquinasa, gonadotropinas; acantocitos; Glut1-metaglut. ** Biomarcadores 2: en ayunas, lactatos, piruvatos, cromatografía de aminoácidos plasmáticos (CAAp), cromatografía de ácidos orgánicos urinarios (UAC), coenzima Q10, ubiquinona.")
Diagrama de flujo del diagnóstico genético de las ataxias.
AMS: atrofia multisistémica; AOA1: ataxia con apraxia oculomotora de tipo 1; AOA2: ataxia con apraxia oculomotora de tipo 2; ARSACS: ataxia espástica autosómica recesiva de Charlevoix-Saguenay; AT: ataxia-telangiectasia; ATN1: atrofina 1; CAG: citosina-adenina-guanina; CANVAS: ataxia cerebelosa, neuropatía, síndrome de arreflexia vestibular; DRPLA: atrofia dentato-rubro-pálido-luysiana; EMG: electromiograma; FRDA: ataxia de Friedreich; IRM: resonancia magnética; NBIA: neurodegeneración con acumulación de hierro en el cerebro; NGS: secuenciación masiva; NOP56: proteína nucleolar 56 (SCA36); NPC1: Niemann-Pick tipo C1; OPA: atrofia óptica; RFC1: subunidad C del factor de replicación (CANVAS); RMF1: retraso mental por cromosoma X frágil 1 (FXS); SCA: ataxia espinocerebelosa; TBP: proteína de unión a caja TATA (SCA17); TC: tomografía computarizada; XCT: cerebrotendinosis xantomatosa.
* Biomarcadores 1: vitamina E, cobre y ceruloplasmina, alfa-fetoproteína, albúmina, hexosaminidasas, ácido fitánico, pristánico, ácidos grasos de cadena muy larga, homocisteína, colesterol, colestanol, Lyso SM509, creatina fosfoquinasa, gonadotropinas; acantocitos; Glut1-metaglut.
** Biomarcadores 2: en ayunas, lactatos, piruvatos, cromatografía de aminoácidos plasmáticos (CAAp), cromatografía de ácidos orgánicos urinarios (UAC), coenzima Q10, ubiquinona.
 e hiperhomocistinemia (homocistinuria tipo III), ácido metilmalónico (MMA) y orótico en orina, pirimidinuria, homocisteína total (tHcy) en plasma (enfermedad de cobalamina C), amonio en plasma (defectos del ciclo de la urea), biotinidasa, fenilcetonuria, hiperglicinemia (encefalopatía por glicina), folato, carnosinuria (homocarnosinosis), lactato, aconitasa, VLCFA (adrenomieloneuropatía), coleterol y triglicéridos, colestanol (XCT), aminoácidos (citrulina, prolina, ornitina, arginina, lisina, cisteína), gonadotropinas, manganeso; aminoácidos, ribitol y D-Arabitol en LCR; trastornos nutricionales: cobre, vitaminas B12, E y β-tocoferol.")
Diagrama de flujo del diagnóstico genético de las paraparesias espásticas hereditarias.
RM: resonancia magnética.
*Trastornos metabólicos: deficiencia de MTHFR (metilentetrahidrofolato reductasa) e hiperhomocistinemia (homocistinuria tipo III), ácido metilmalónico (MMA) y orótico en orina, pirimidinuria, homocisteína total (tHcy) en plasma (enfermedad de cobalamina C), amonio en plasma (defectos del ciclo de la urea), biotinidasa, fenilcetonuria, hiperglicinemia (encefalopatía por glicina), folato, carnosinuria (homocarnosinosis), lactato, aconitasa, VLCFA (adrenomieloneuropatía), coleterol y triglicéridos, colestanol (XCT), aminoácidos (citrulina, prolina, ornitina, arginina, lisina, cisteína), gonadotropinas, manganeso; aminoácidos, ribitol y D-Arabitol en LCR; trastornos nutricionales: cobre, vitaminas B12, E y β-tocoferol.

Con este conjunto mínimo de pruebas diagnósticas, y si no se ha podido llegar a un primer diagnóstico de certeza, pero la sospecha clínica es clara, el paciente también puede beneficiarse de una derivación a un centro de referencia en la atención de ataxias y paraparesias espásticas hereditarias (CSUR). En la actualidad se dispone de siete centros acreditados en nuestro país: Hospital Universitario Marqués de Valdecilla (Cantabria), Hospital Universitari Vall d’Hebron, Hospital Clinic, Hospital Sant Joan de Deu (Cataluña), Hospital Universitario La Paz, Hospital Universitario Ramón y Cajal, y el Hospital Universitario y Politécnico La Fe (Valencia).
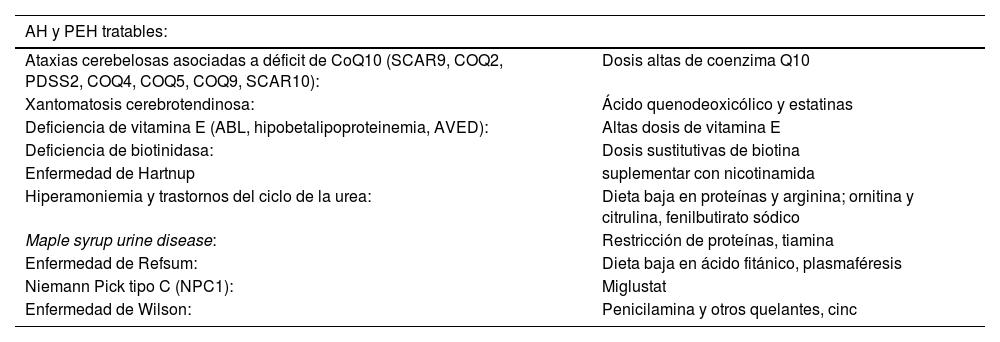
Tratamiento y pronósticoAlgunas ataxias y paraparesias espásticas hereditarias son tratables etiológicamente por lo que es importante identificarlas (tabla 3)20,23,26.
Ataxias y paraparesias espásticas con tratamiento específico disponible
| AH y PEH tratables: | |
|---|---|
| Ataxias cerebelosas asociadas a déficit de CoQ10 (SCAR9, COQ2, PDSS2, COQ4, COQ5, COQ9, SCAR10): | Dosis altas de coenzima Q10 |
| Xantomatosis cerebrotendinosa: | Ácido quenodeoxicólico y estatinas |
| Deficiencia de vitamina E (ABL, hipobetalipoproteinemia, AVED): | Altas dosis de vitamina E |
| Deficiencia de biotinidasa: | Dosis sustitutivas de biotina |
| Enfermedad de Hartnup | suplementar con nicotinamida |
| Hiperamoniemia y trastornos del ciclo de la urea: | Dieta baja en proteínas y arginina; ornitina y citrulina, fenilbutirato sódico |
| Maple syrup urine disease: | Restricción de proteínas, tiamina |
| Enfermedad de Refsum: | Dieta baja en ácido fitánico, plasmaféresis |
| Niemann Pick tipo C (NPC1): | Miglustat |
| Enfermedad de Wilson: | Penicilamina y otros quelantes, cinc |
Sin embargo, la mayoría de los tipos de AH y PEH se caracterizan por una evolución progresiva con cada vez mayor discapacidad funcional. Por tanto, los esfuerzos deben ir encaminados a intentar mantener la situación funcional mediante terapias rehabilitadoras como la fisioterapia, la logopedia y la terapia ocupacional. Es importante considerar la necesidad de ortesis y dispositivos para la deambulación como bastones, andadores y silla de ruedas. Reconocer el grado de discapacidad permite identificar y adecuar los recursos necesarios para estos pacientes.
Por otro lado, los tratamientos farmacológicos sintomáticos ayudarán a mejorar la espasticidad y favorecer la deambulación y la corrección de posturas con infiltraciones de toxina botulínica o tratamientos orales o intratecales como baclofeno y tizanidina. También, la oxibutinina es útil para mejorar la urgencia miccional. Las aminopiridinas (4-AP, fampridina o dalfampridina) pueden ser útiles para el downbeat nystagmus, y acetazolamida y/o 4-AP para la ataxia episódica tipo 2 (EA2)23–26.
Finalmente, se deben tener en cuenta medidas de adaptación en domicilio y los recursos sociales disponibles por Comunidades Autónomas o a nivel nacional, y Ley de Dependencia. Hay que tener en cuenta el carácter lentamente progresivo con la consiguiente variación el grado de discapacidad y la calidad de vida a lo largo de la enfermedad.
Discusión y conclusionesLa falta de un diagnóstico y de una valoración del grado de discapacidad adecuado en este grupo de enfermedades conlleva para el paciente y sus familiares directos no solo la incertidumbre de convivir con un diagnóstico incierto, sino que, además, puede limitar los recursos y opciones sociales de los que se podrían beneficiar.
El objetivo de este trabajo es la de mejorar el diagnóstico y facilitar y homogeneizar la evaluación de la discapacidad de los pacientes con AH y PEH. Es fundamental que haya una valoración equitativa independientemente del equipo médico (neurólogo especializado o no, médicos de primaria, rehabilitadores, peritos médicos…) y la Comunidad Autónoma donde se haga el proceso.
En la literatura hay publicadas algunas guías de discapacidad globales en enfermedades raras3,4 o en enfermedades más frecuentes como el ictus o la enfermedad de Parkinson, pero no se ha publicado hasta la fecha ninguna específica relacionada con AH y PEH. Deseamos que este documento, junto con su versión ampliada disponible en la web de la Sociedad Española de Neurología facilite la asistencia clínica diaria del neurólogo general y otros médicos especialistas como médicos rehabilitadores, internistas, médicos de Atención Primaria, o Medicina del Trabajo.
Conclusiones: las AH y PEH son enfermedades neurodegenerativas poco frecuentes que siguen en la mayor parte de los casos un curso lentamente progresivo con cada vez mayor repercusión funcional. La realización de un estudio sistemático y común para todos los pacientes en los que se sospeche una AH o PEH puede mejorar su diagnóstico y adecuada valoración. Con esta guía se pretende facilitar la asistencia clínica diaria del neurólogo general y de los médicos que valoran la discapacidad.
Conflicto de interesesLos autores declaran no tener ningúnconflicto de intereses.

