



La hemorragia subaracnoidea de la convexidad cerebral (HSAc) se caracteriza por colecciones hemáticas en uno o varios surcos adyacentes, sin sangrado en el parénquima cerebral, cisura interhemisférica, cisternas basales o ventrículos1–3. Se ha asociado a múltiples etiologías como traumatismos, enfermedad aneurismática, oclusión de venas corticales, síndrome de leucoencefalopatía posterior reversible, síndrome de vasoconstricción cerebral reversible (SVCR), angiopatía amiloide cerebral (AAC), vasculitis primaria del sistema nervioso central, coagulopatías, consumo de cocaína o etanol, abscesos cerebrales, cavernomas o malformaciones arteriovenosas1–4. A continuación presentamos un caso de HSAc como manifestación de telangiectasia hereditaria hemorrágica (THH).
Varón de 47 años con antecedente de epistaxis repetidas desde la infancia e historia familiar de epistaxis en la madre, tío y abuelo maternos, quienes requirieron cauterización de telangiectasias en la cavidad nasal. Refería una cefalea opresiva frontal izquierda, de intensidad y frecuencia crecientes (hasta hacerse diaria) en los últimos 5 meses. Esta aumentaba con la maniobra de Valsalva, asociando vértigo y vómitos, con fotopsias ocasionales. No refería fiebre, consumo de sustancias vasoactivas o transgresión etílica. A pesar del dexketoprofeno intravenoso 50mg/8h, tramadol/paracetamol oral 37,5/325mg/8h y prednisona oral 60mg/24h, precisó rescates de cloruro mórfico subcutáneo a dosis de 5mg.
La exploración sistémica destacó un angioma cutáneo en ápice nasal y puntos rubí en lengua y labio inferior; la exploración neurológica fue normal.
Los estudios de coagulación, hemograma y bioquímica fueron normales. El líquido cefalorraquídeo mostró composición y presión normales, sin hematíes en el mismo. La secuenciación de exoma mostró una mutación con cambio de sentido en el exón 3 del gen del receptor de activina A tipo II-1, ACVRL1 (c.236G>A; p.(Gly79Glu)), en el cromosoma 12, no descrita anteriormente.
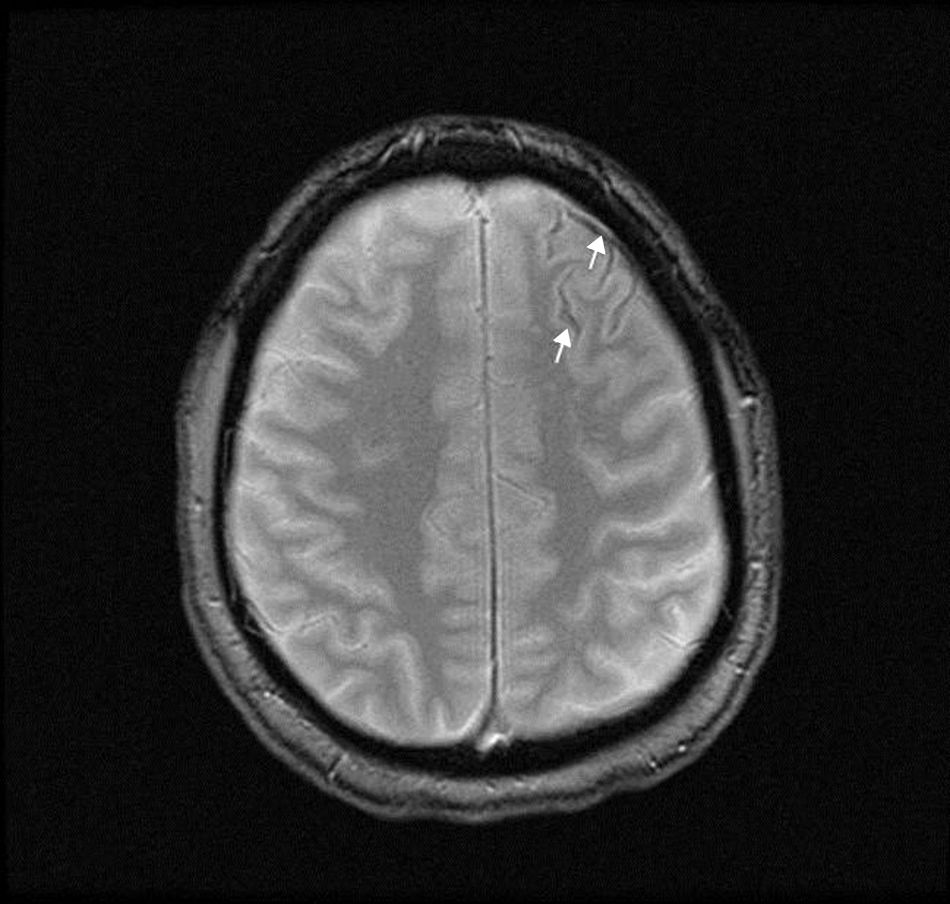
Una RM cerebral, en secuencia T2, demostró un fino ribeteado de artefacto de susceptibilidad magnética en los surcos frontales izquierdos compatible con depósitos de hemosiderina (fig. 1). La administración de gadolinio no mostró realces patológicos ni hallazgos sugerentes de inflamación de paredes vasculares. Un dúplex transcraneal y una arteriografía cerebral fueron normales. Mediante rinoscopia se demostraron telangiectasias en el tabique nasal y los cornetes, que se cauterizaron.
 en los surcos de la convexidad frontal izquierda, correspondientes con HSAc.")
La THH, conocida como enfermedad de Rendu-Osler-Weber es una enfermedad de herencia autosómica dominante, con una prevalencia de entre 1:5.000 y 1:8.0005–7. La epistaxis es la manifestación más frecuente, y las telangiectasias mucocutáneas, el signo más común.
El diagnóstico clínico se realiza mediante los criterios de Curaçao8, basados en la presencia de: 1) epistaxis recurrente y espontánea; 2) telangiectasias mucocutáneas múltiples en localizaciones características (labios, lengua, mucosa oral y punta de los dedos); 3) participación visceral (gastrointestinales, pulmonares, hepáticas o cerebrales9), y 4) un familiar de primer grado con THH. El diagnóstico es definitivo, si se cumplen al menos 3 de los criterios enumerados8.
Cuando existe sospecha diagnóstica, o en ausencia de antecedente familiar, se recomienda la realización de un estudio genético9; Además de presentar los 4 criterios clínicos, este paciente presentó una variante genética en ACVRL1, cuyas mutaciones patogénicas se han asociado siempre a THH10.
La THH causa numerosas manifestaciones neurológicas, como malformaciones vasculares cerebrales, abscesos, ictus isquémicos, crisis epilépticas y migraña. Entre las primeras destacan malformaciones arteriovenosas (MAV) múltiples, silentes y de bajo flujo, presentes en el 10% de los casos, que pueden causar hemorragias devastadoras10–12. Otras son telangiectasias capilares, malformaciones cavernosas, anomalías del desarrollo venoso, fístulas arteriovenosas y aneurismas saculares13. Los ictus isquémicos se presentan en un 30% de pacientes con MAV pulmonares, por un mecanismo de embolismo paradójico14.
La etiología de HSAc es variada y difiere en función de la edad de presentación, de forma que en menores de 60 años predomina el SVCR, y en mayores de 60 años la principal causa es la AAC1–4. La sintomatología también varía según la edad: la cefalea en trueno es el síntoma más frecuente en el primer grupo etario, y los episodios focales transitorios conocidos como «amyloid spells», en el segundo1–3. Se considera que estos episodios podrían estar causados por un mecanismo de depresión cortical propagada similar al de la migraña, secundario a la presencia de sangre en la corteza cerebral3. La TC cerebral sin contraste es la prueba diagnóstica más inmediata, si bien su sensibilidad es limitada, por lo que la RM cerebral resulta necesaria para el diagnóstico15. En este caso, la RM mostró depósitos de hemosiderina en los surcos corticales frontales anteriores, indicando sangrado previo compatible con HSAc, mientras que la arteriografía fue normal. Probablemente, este paciente presentase una malformación vascular causante del sangrado que, debido a rotura, tamaño, bajo flujo o vasoespasmo, no fuera apreciable en las pruebas de imagen.
En resumen, la THH puede ser causa de HSAc, hecho no descrito previamente en la literatura médica. Es también de interés el hallazgo de una nueva variante genética en ACVRL1, cuyo significado patogénico deberá determinarse mediante los estudios de segregación familiar oportunos.

