



La lisencefalia es una alteración de la corteza cerebral que se produce por un defecto en la migración y desarrollo de las neuronas del sistema nervioso1,2. Las neuronas al no poder migrar hasta las capas superficiales de la corteza cerebral generan una disminución de las circunvoluciones cerebrales y se acumulan en capas profundas de sustancia blanca formando una banda subcortical (heterotopía en banda). Existen 2 tipos clásicamente descritos de esta entidad. El tipo 1 en el que se puede dar una ausencia completa de circunvoluciones (agiria) que haga que la superficie cerebral sea lisa o en los que se produzca una formación incompleta que genere un aumento del tamaño de las circunvoluciones que persistan (paquigiria). En el tipo 2 predominan cúmulos desorganizados de neuronas con una orientación desordenada, sin formar capas definitivas ni seguir un patrón predecible. Este último tipo se ha asociado a varios síndromes genéticos como la distrofia muscular de Fukuyama, la enfermedad músculo-ojo-cerebro de Santavuori o el síndrome de Meckel-Gruber3. Así mismo, la lisencefalia se puede asociar a otras malformaciones tanto del sistema nervioso como a otros niveles del organismo entre las que destaca su asociación con la hipoplasia cerebelar (LHC)1,2,4–6, que se puede observar en otros síndromes genéticos como el síndrome Walker-Warburg3. Con respecto a la LCH se han descrito mutaciones en los genes: RELN y TUBA1A. El gen RELN, localizado en brazo largo de cromosoma 7 (7q22) codifica la proteína reelina que participa en la migración neuronal, la plasticidad sináptica y la transmisión de los impulsos nerviosos. Se conocen al menos 6 mutaciones que se heredan con patrón autosómico recesivo. El TUBA1A, situado en brazo largo de cromosoma 12 (12q13.12) codifica la proteína alfa-tubulina, que forma parte de los microtúbulos intracelulares cuya función consiste en la división y movimiento celular. Se han descrito 10 mutaciones con patrón de herencia autosómico dominante y se han encontrado en un 30% de los casos de LCH1.
En cuanto a la atresia de vías biliares extrahepática (ABEH) se clasifican en 3 tipos dependiendo de si se presenta como única anomalía (tipo I), asociada a otros defectos congénitos, pero sin considerarse un síndrome polimalformativo (tipo II) o si forma parte de este (tipo III)7,8. En los tipos II y III se describen anomalías cardíacas, gastrointestinales, esplénicas y genitourinarias.
En el caso que presentamos fue especialmente complicado el manejo de las crisis neonatales secundarias al trastorno de desarrollo de la corteza cerebral y posteriormente por la situación de insuficiencia hepática con desenlace fatal. Por el momento no se han descrito asociación ni mutación genética que asocie LCH y ABEH como el caso que presentamos a continuación.
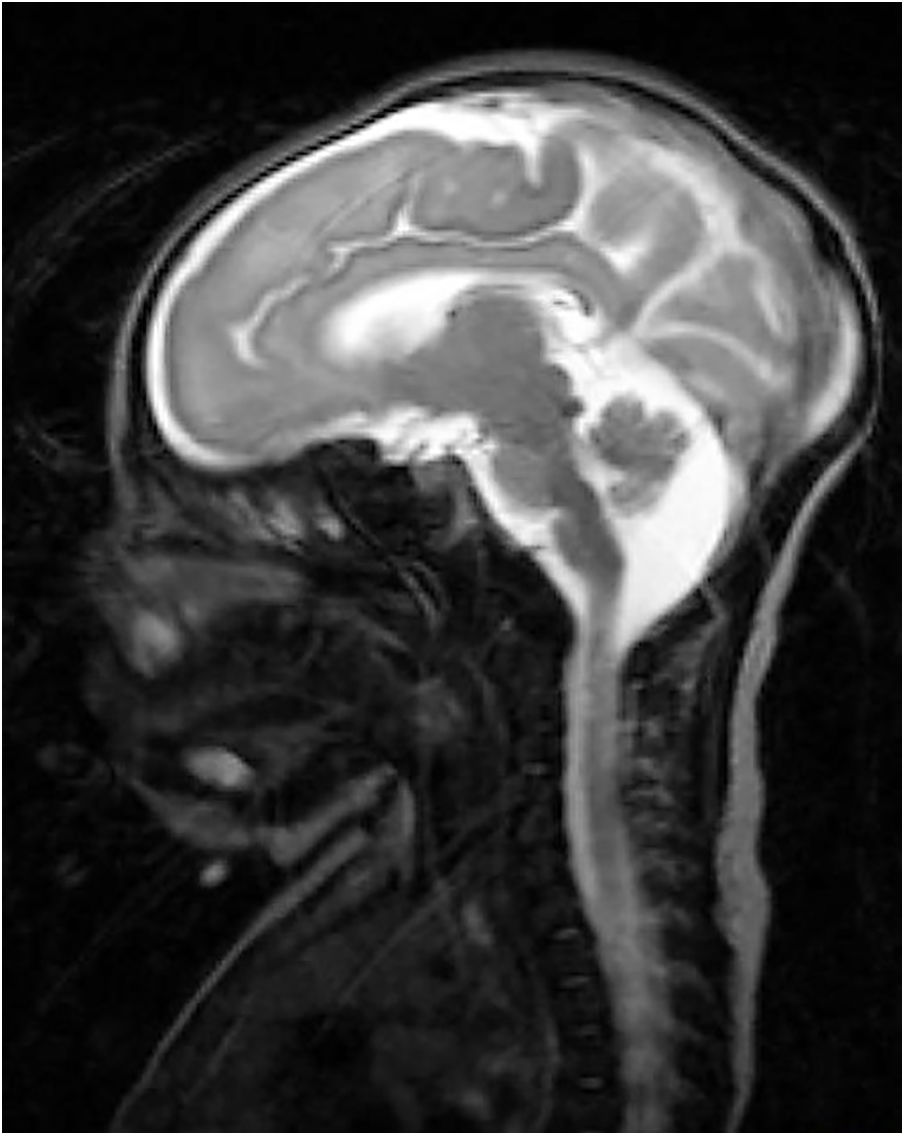
Recién nacido a término con retraso del crecimiento intrauterino a partir de la semana 34. Sin antecedentes gestacionales de interés. No consanguinidad paterna. Parto eutócico. Peso al nacer: 2.470g (p 2, DE: −2,18), talla: 46cm (p 2, DE: −2,3) y perímetro craneal: 30cm (<p 1, DE: −3,21). A las 2h de vida comienza con movimientos de chupeteo que posteriormente se asocian con movimientos clónicos de cara y miembro superior izquierdo de 4min de duración. Presenta episodios similares durante las primeras 24h de vida que ceden con tratamiento con fenobarbital. Se realiza estudio de convulsión neonatal en el que destaca una trombocitosis y un ligero aumento de hormonas tiroideas en analítica sanguínea. Se realiza un electroencefalograma que es normal. En ecografía transfontanelar, se observa un adelgazamiento de cuerpo calloso con una megacisterna magna. Se realiza resonancia magnética cerebral (fig. 1) que describe, además de los hallazgos observados previamente, atrofia parcial de vermis y moderada disminución de la surcación a nivel frontal y occipital compatible con LCH. Requiere tratamiento con fenobarbital, levetiracetam y clobazam por mal control clínico de epilepsia. Al mes y medio de vida se observa colestasis en analítica de control (bilirrubina total de 6,8mg/dl, directa de 5,3mg/dl y gamma-glutamil transferasa de 2.120UI/l). Se amplía estudio de colestasis con estudio alfa-1-atripsina, infeccioso, hormonal, metabólico y ecografía abdominal que son normales. Ante la sospecha de ABEH se decide realizar tratamiento conservador por mal pronóstico de la paciente. Se realiza estudio genético de lisencefalia y defectos de la glicosilación que resultan negativos. A los 7 meses, consulta por fiebre y descompensación de su hepatopatía, pero a pesar del tratamiento médico, fallece por parada cardiorrespiratoria. En autopsia, se confirma la ABEH gracias a la biopsia hepática. Es el primer caso descrito en la literatura de LHC con ABEH. No existen estudios ni se han descubierto mutaciones genéticas que relacionen la LCH con ABEH.
Financiación
No existen fuentes de financiación.

