



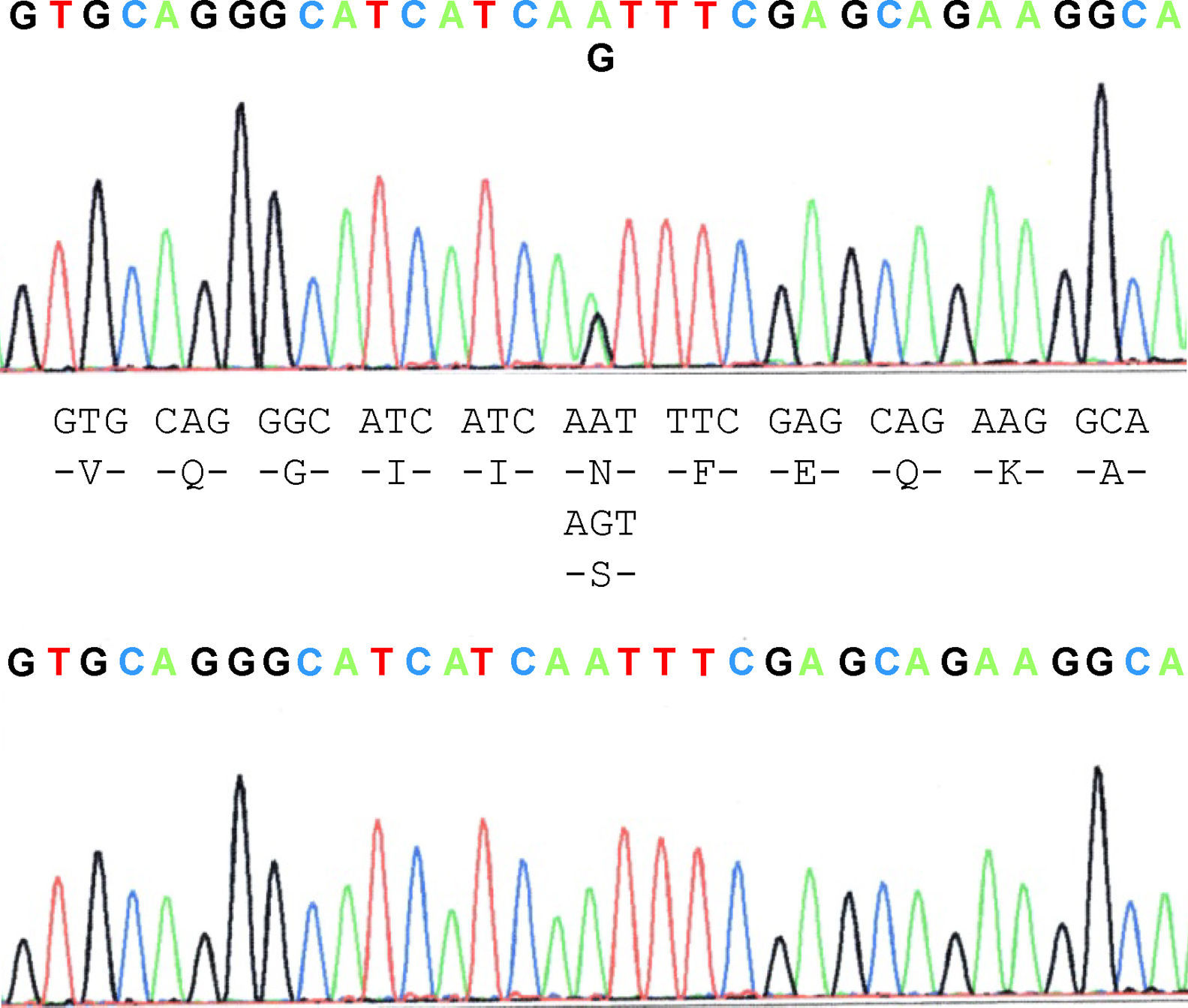
La N19S es una mutación por la sustitución en la posición 139 de la SOD1 y fue descrita por Mayeux et al, donde los autores sugirieron que no tenía un efecto causal al hallarse casos asintomáticos y esporádicos pero autores posteriores han sugerido lo contrario.
Material y métodosSe describe una familia con 4 pacientes con ELA en los que en el caso propósito es portador de la mutación N19S. Se realiza un estudio molecular en 15 personas de la familia de diferente orden.
ResultadosLa enfermedad se halla en la línea materna del primer caso. Se detecta la presencia de la mutación en tres personas, el primer y dos asintomáticos. Uno de los pacientes afectos de ELA de la familia que murió previamente, no presentaba la mutación. Dos de los hijos del tercer caso y otro del cuarto caso tampoco la mostraron. Contrariamente, la mutación está presente en la rama paterna del primer caso, que es asintomática.
ConclusiónLa familia descrita apoya la hipótesis de Mayeux et al y va en contra que la mutación N19S tenga connotaciones patológicas, ya que la mutación se encuentra en la línea familiar no afecta de la enfermedad y no está en la línea con enfermos de ELA. En consecuencia, aunque el caso descrito es una forma familiar no puede ser atribuido a la mutación y su relación debe ser considerada como casual.
N19S mutation is produced by substitution in the 139 position of SOD1 and was described by Mayeux in a patient with amyotrophic lateral sclerosis (ALS). He suggested that it did not have a causal effect as it was found in asymptomatic and sporadic cases. Other authors in later articles did not agree.
Material and methodsWe describe a family with 4 members with ALS patients and attempt to find the carrier of the N19S mutation of the propositus. Molecular studies were performed on 15 members of the family of a different order.
ResultsThe ALS cases were found in the maternal line of the propositus. The presence of the mutation was detected in 3 people, the other two were asymptomatic. One of patients with ALS in the family, who died previously, did not have the mutation. Two of the sons of this case and another of the other case did not show it. On the other hand, N19S mutation was only present in paternal branch of the propositus, where there were no cases.
ConclusionThe described family supports the hypothesis by Mayeux and against that mutation N19S has pathological consequences, since mutation is only in the family line where there are no cases with ALS. In consequence, although the described case is included as a familiar form, it cannot be attributed to the mutation, and its relationship with N19S should be considered as casual.
Un 10% de los casos de esclerosis lateral amiotrófica (ELA) son familiares1 y de ellos, aproximadamente un 25% están asociados a una mutación en el gen de la superoxido-dismutasa-1 (SOD-1)2, de las que se han descrito más de 100 tipos diferentes3,4. La producción de una proteína SOD-1 anómala por estas mutaciones podría precipitar los mecanismos de la apoptosis y la lesión neuronal5 y a través de alguna de ellas se obtuvieron modelo experimentales transgénicos de ELA familiares6,7 lo que ha permitido postular líneas terapéuticas8, aunque existe la controversia que puedan ser aplicables a los pacientes esporádicos9,10. Aunque la mayoría de las formas familiares descritas asociadas a mutaciones SOD1 son dominantes11 algunas de ellas se han observado con una herencia recesiva12 y por ello se han descrito pacientes que podrían ser esporádicos13–16 o formas heterocigóticas de penetrancia incompleta17,18, pero también podría sugerirse que pudieran ser asintomáticos o no tener significación patológica. La mutación N19S se produce por la sustitución en la posición 139, de ASN por SER, en exón 1 y su primera descripción se debe a Mayeux et al19 en 2003 y consideró que podría ser asintomática. En el presente artículo presentamos un caso en una familia de ELA que mostraba la mutación N19S.
Material y métodosCaso propósitoLa paciente estudiada presentaba 52 años en el momento del diagnóstico sin antecedentes médicos de interés, comenzó a mostrar debilidad y amiotrofia en los segmentos distales del miembro superior derecho de manera progresiva. En la exploración neurológica inicial además de paresia y atrofia de los músculos distales, se observaron fasciculaciones e hiperreflexia en la extremidad afectada. No presentaba déficits sensitivos ni otros signos neurológicos asociados. El electromiograma (EMG) objetivó fasciculaciones y actividad espontánea de denervación, así como pérdida de unidades motoras en los trazados voluntarios de máximo esfuerzo en varios músculos del miembro superior derecho. Por lo demás, en el estudio neurofisiológico no se detectaron bloqueos de conducción y las velocidades de conducción nerviosa se hallaban dentro de rangos normales. Se realizó resonancia magnética (RM) de cerebro y de columna cervical, sin encontrar ninguna lesión estructural. Otras pruebas complementarias fueron asimismo normales, incluyendo hemograma y perfil bioquímico de sangre, inmunoelectroforesis, hormonas tiroideas, niveles de vitamina B12, serologías de Borrelia, sífilis y VIH, anticuerpos antinucleares, antigangliósido y antineuronales, así como el estudio citobioquímico del líquido cefalorraquídeo. Desde las primeras fases del cuadro clínico, se prescribió riluzol a una dosis de 100mg al día. Los síntomas y signos motores progresaron y se extendieron hacia músculos proximales de forma paulatina, aunque han progresado lentamente. Finalmente, tres años después del comienzo, las manifestaciones clínicas se extendieron a las restantes extremidades. En ese momento la exploración neurológica reveló la presencia de signos motores bilaterales, con debilidad, fasciculaciones y atrofia muscular en ambos miembros superiores, fasciculaciones en ambos cuádriceps femorales e hiperreflexia universal. En estas etapas, el EMG mostraba ya un patrón neurogénico crónico en los músculos de las cuatro extremidades. La musculatura bulbar no está todavía afectada.
Estudio familiarLa paciente tenía antecedentes familiares de la enfermedad por línea materna, uno de sus tíos y los dos hijos del mismo habían sufrido una ELA con criterios clínicos. La línea paterna de esta familia está compuesta por 6 personas de primer orden y 18 de segundo orden. La línea materna está constituida por 9 personas de primer orden y 19 de segundo orden, a añadir el caso propósito y sus 5 hermanos. De los 58 miembros de la familia entre primer y segundo orden se han estudiado 12 personas, además de tres más de tercer orden.
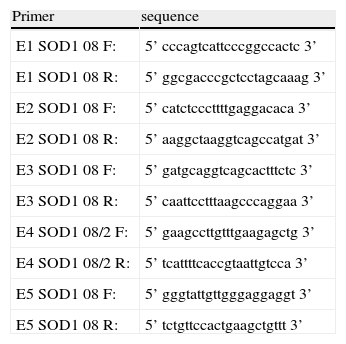
Análisis molecularLas muestras sanguíneas del paciente, familiares y controles se han obtenido con consentimiento informado. La extracción de DNA se ha realizado a través de procedimientos estandarizados utilizando una PCR para ampliar los cinco exones del gen de la SOD1 (tabla 1). La detección de las variaciones en las secuencias se ha realizado usando secuenciación. Los productos de la PCR fueron purificados con el GFX PCR DNA purification kit (Amersham Pharmacia Biotech, Piscataway, New Jersey). La secuenciación directa de los productos de PCR fue realizada con los mismos set de primers (anteverso e inverso) en un ABIPrism 310 DNA sequenciador y el cycle dye terminator DNA sequencing kit de Applied Biosystems (Perkin Elmer–Applied Biosystems, Foster City, California). Los análisis fueron realizados mediante un software de análisis de secuencia con secuencia de referencias del GenBank L44135 a L44139.
Lista de primers para obtener los 5 productos de PCR correspondientes a los 5 exones SOD1 y sus flanking 5’ and 3’ secuencias
| Primer | sequence |
| E1 SOD1 08 F: | 5’ cccagtcattcccggccactc 3’ |
| E1 SOD1 08 R: | 5’ ggcgacccgctcctagcaaag 3’ |
| E2 SOD1 08 F: | 5’ catctcccttttgaggacaca 3’ |
| E2 SOD1 08 R: | 5’ aaggctaaggtcagccatgat 3’ |
| E3 SOD1 08 F: | 5’ gatgcaggtcagcactttctc 3’ |
| E3 SOD1 08 R: | 5’ caattcctttaagcccaggaa 3’ |
| E4 SOD1 08/2 F: | 5’ gaagccttgtttgaagagctg 3’ |
| E4 SOD1 08/2 R: | 5’ tcattttcaccgtaattgtcca 3’ |
| E5 SOD1 08 F: | 5’ gggtattgttgggaggaggt 3’ |
| E5 SOD1 08 R: | 5’ tctgttccactgaagctgttt 3’ |
La paciente propósito que cumplía los criterios de ELA20 mostró ser portadora de la mutación N19S (fig. 1). De los miembros de la familia estudiados, tres de ellos, la paciente y dos personas más asintomáticas, el padre de 87 años y un hermano de 48 años, eran portadores de la mutación. Aparte de ella, en el material biológico de uno de los pacientes afectos de ELA de la familia (caso 3) murió previamente y estaba archivado, pudo constatarse que no presentaba la mutación. Dos de los 5 hijos del caso 3, no mostraron la presencia de la mutación ni uno de los tres hijos del caso 4. Contrariamente a lo esperado, el estudio mostró que la mutación está presente en la rama paterna del caso propósito, es decir, aquella que no tiene casos de ELA. Ello, sugiere que la forma familiar de ELA no está afecta de la mutación, salvo en el propósito, que es la línea materna, mientras que la mutación se halla en la línea paterna donde no hay pacientes, sugiriendo que la enfermedad podría no estar relacionada con la mutación y su presencia sería casual.
Discusión
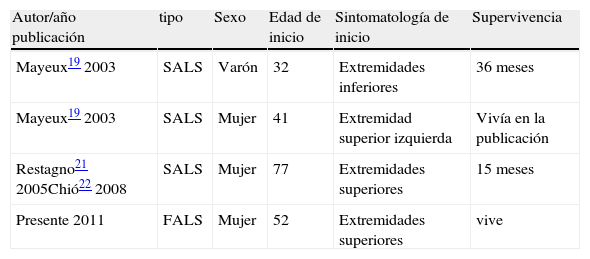
La mutación N19S se descrbió por Mayeux et al19 y posteriormente se han descrito algunos casos más21,22 pero no en formas familiares (tabla 2). En el caso original, se describían siete familiares de quince estudiados en tres generaciones (entre 30 y 80 años) que presentaban la mutación sin que tuvieran clínica, lo que llevó a los autores a sugerir que la asociación a la mutación no suponía la causa de la enfermedad, ya que además encontraron un sujeto con la mutación entre 180 controles.
Casos descritos con la asociación de ELA a N19S
| Autor/año publicación | tipo | Sexo | Edad de inicio | Sintomatología de inicio | Supervivencia |
| Mayeux19 2003 | SALS | Varón | 32 | Extremidades inferiores | 36 meses |
| Mayeux19 2003 | SALS | Mujer | 41 | Extremidad superior izquierda | Vivía en la publicación |
| Restagno21 2005Chió22 2008 | SALS | Mujer | 77 | Extremidades superiores | 15 meses |
| Presente 2011 | FALS | Mujer | 52 | Extremidades superiores | vive |
El papel de las mutaciones SOD1 en la ELA está en discusión10,23. Se ha señalado la posibilidad de que una misma mutación tenga una penetrancia diferente y el ejemplo más característico es la mutación SOD1-D90A24,25. Esta mutación tiene dos formas fenotípicas, una homocigótica, que se describe en Torne Valley, entre Suecia y Finlandia, y una forma heterocigótica, que se encuentra en otros países europeos26. Las formas homocigotas son benignas, caracterizadas por piramidalismo y larga supervivencia, mientras que las formas heterocigóticas son mucho más agresivas y similares a las formas familiares dominantes portadoras de otras mutaciones SOD127. En este sentido la mutación SOD1-N19S también podría presentar fenotipos diferentes. Otra posibilidad que puede explicar un fenotipo distinto con una misma mutación podría relacionarse por una potencial disminución de la carga de la mutación como se ha demostrado en el ratón transgénico a medida que pasan las generaciones, donde la enfermedad se modifica e incluso aumenta su supervivencia28.
En la publicación original en que se describía la mutación N19S, Mayeux et al19 sugerían que la asociación no era causal, dado que la sustitución de AA ocurre en un lugar de la molécula con poca relevancia, ya que para estos autores, N19 no está conservado en diferentes especies y por tanto no debía tener un papel relevante en la función de SOD1 y su sustitución solo podría generar modificaciones menores. Sin embargo, Obata et al29 han realizado un estudio experimental con la mutación donde trasfectaron cDNA-N19S-SOD1 a células NSC34, que son células hibridas de motoneuronas embrionarias de medula espinal procedentes de raton y células procedentes de neuroblastoma de ratón hallando que la sobre-expresión de N19S-SOD1 inducía muerte neuronal en estas células, que se producían agregados intracelulados en baja proporción (mayor que controles, pero menor que las mutaciones SOD1 más frecuentes), que se mostraba estres oxidativo que, mediante tratamiento oxidativo, aumentaba la polimerización, aunque en menor grado que otras mutaciones, con poliubiquitinización acelerada, de forma similar a otras mutaciones SOD1, concluyendo que la mutación N19S producía muerte de la motoneurona y generaba una proteína mutada lo que les sugería que producía la enfermedad en probablemente en combinación con otros factores de riesgo.
Nuestra familia, por el contrario apoya la hipótesis de Mayeux et al19 y va en contra que tenga connotaciones patológicas ya que su presencia se encuentra en la línea familiar no afecta de la enfermedad y no está en la línea con enfermos de ELA. Se trata de una situación parecida a la descrita por Flebecker et al para las mutaciones D90A y E100K, donde halla pacientes con ELA con y sin mutaciones en el seno de una misma familia30. En consecuencia, aunque nuestro caso es una forma familiar no puede ser atribuido a la mutación y su relación debe ser considerada como casual y confirma que la asociación a una mutación del gen de la SOD puede ser asintomática31.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

