



La polineuropatía amiloidótica familiar por transtiretina (PAF por TTR) cursa inicialmente con una afectación preferente de fibra fina e importante afectación autonómica, con un inicio en la segunda-tercera década de la vida y una evolución rápidamente progresiva que puede llevar a la muerte del paciente en unos 10 años. Sin embargo, en zonas no endémicas, se han descrito otros fenotipos.
Objetivos y métodosDescribimos 4 casos procedentes de una zona no endémica, la provincia de Guipúzcoa, con el objetivo de mostrar la variabilidad clínica de esta enfermedad.
Pacientes y resultadosTres pacientes presentaron una forma de inicio tardío, por encima de los 50 años, que cursó como una polineuropatía de predominio motor con afectación distal inicial en miembros inferiores y posteriormente en los superiores. Uno sufrió dolor neuropático intenso. Ninguno tuvo signos de afectación autonómica. La cuarta paciente, de origen portugués, presentó una forma típica de inicio en la treintena, con dolor neuropático y disautonomía. Los 4 pacientes presentan la mutación Val50Met en el gen TTR.
ConclusiónLa PAF es una enfermedad pleomórfica incluso en pacientes con la misma mutación. En zonas no endémicas, su presentación predominante puede ser como una polineuropatía de inicio por encima de la sexta década, de predominio motor y sin signos disautonómicos. Dado lo inespecífico de esta forma de presentación y la facilidad técnica con la que se puede estudiar actualmente el gen TTR, creemos que en el protocolo de diagnóstico etiológico de cualquier polineuropatía se debe incluir la secuenciación de este gen.
Transthyretin-related familial amyloid polyneuropathy (TTR-FAP) typically arises as an autonomic neuropathy primarily affecting small fibres and it occurs in adult patients in their second or third decades of life. It progresses rapidly and can lead to death in approximately 10 years. Other phenotypes have been described in non-endemic areas.
Objectives and methodsWe described 4 cases from the Spanish province of Guipuzcoa, a non-endemic area, to highlight the clinical variability of this disease.
Patients and resultsThree patients presented a late-onset form manifesting after the age of 50, featuring a predominantly motor polyneuropathy initially causing distal impairment of the lower limbs followed by the upper limbs. One patient suffered severe neuropathic pain. None showed signs of autonomic involvement. The fourth patient, of Portuguese descent, presented a typical form with onset in her thirties, neuropathic pain and dysautonomia. All patients carry the Val50Met mutation in the TTR gene.
ConclusionFAP is a pleomorphic disease even in patients carrying the same mutation. In non-endemic areas, its main form of presentation may resemble a predominantly motor polyneuropathy developing in the sixth decade of life with no signs of dysautonomia. Given this non-specific presentation and the widely available technical means of studying the TTR gene, we believe that the protocol for the aetiological diagnosis of any polyneuropathy should include genetic sequencing of TTR.
La polineuropatía amiloidótica familiar por transtiretina (PAF por TTR) es una enfermedad multisistémica de base genética producida por mutación en el gen de la TTR1-7. Fue descrita en 1952 por Corinho Andrade en pacientes del norte de Portugal2,3,8, donde se conocía popularmente como mal dos peshinos5.
Existen 3 focos endémicos a nivel mundial que incluyen Portugal, Suecia y Japón2,4,5,8,9. En España los focos endémicos más importantes se sitúan en la isla de Mallorca7,8,9 y en la provincia de Huelva (Valverde del Camino)7. El País Vasco es una zona no endémica. A nivel mundial se estima que tiene una prevalencia de 1/10.000 habitantes1 y en Europa de 1/100.000 habitantes7,8.
El gen de la TTR es de pequeño tamaño. Consta solo de 4 exones y está localizado en el brazo corto del cromosoma 18 (18q12.1)5. Se han descrito más de 100 mutaciones asociadas a este gen. La más frecuente es la que provoca una sustitución del aminoácido valina por metionina en la posición 30 (Val30Met o V30M, ahora denominada Val50Met debido a una actualización de la secuencia de referencia que ha cambiado el orden de los aminoácidos) de los 127 aminoácidos que constituyen la proteína1-8.
La enfermedad se transmite de forma mayoritaria con un patrón de herencia autosómico dominante7,8, aunque existen casos esporádicos y mutaciones de novo5. La penetrancia es muy variable, diferente de una mutación a otra e incluso, para la misma mutación, distinta según el foco endémico5-8. Se ha descrito una anticipación en la edad de aparición de las manifestaciones clínicas y una mayor penetrancia cuando el transmisor de la mutación es una mujer5,8.
La TTR es una proteína precursora del amiloide que actúa como transportador de la tiroxina y de la vitamina A1,5-8. Se sintetiza de forma mayoritaria en el hígado (95%), y en un pequeño porcentaje en la retina y en los plexos coroideos. Tanto en la sangre como en el líquido cefalorraquídeo circula en forma de un complejo tetramérico. La TTR mutada hace que se desestabilice el tetrámero, dando lugar a monómeros que se pliegan anormalmente y precipitan de forma parcheada en los diferentes tejidos, principalmente en el sistema nervioso periférico y corazón1,5-7.
La variabilidad en cuánto a edad de inicio y presentación clínica de esta patología, sobre todo en zonas no endémicas, dificulta y retrasa su diagnóstico.
Pacientes, métodos y resultadosSe describen 4 pacientes identificados en la consulta de neurología del Hospital Universitario Donostia entre 2005 y 2016, que muestran la variabilidad clínica de esta patología.
Paciente 1Varón de 58 años de origen navarro que acudió a consultas de neurología en diciembre de 2013 refiriendo cansancio y sensación «rara» en los pies de un año de evolución. En los últimos meses, asociaba una sensación similar en las manos, presentando dificultad para realizar algunos movimientos finos, como abrir una pinza. No refería síntomas de disautonomía.
No presentaba antecedentes de interés salvo ambliopía del ojo izquierdo desde la infancia e intervención de desprendimiento de retina y cataratas. La exploración neurológica era normal, salvo por una leve debilidad para la dorsiflexión de tobillos y una discreta alteración sensitiva distal en «calcetines».
La analítica general y el sedimento de orina (que incluía microalbúmina) fueron normales. El estudio electromiográfico del músculo abductor corto del pulgar y del músculo tibial anterior izquierdo no mostraron alteraciones. El estudio electroneurográfico fue compatible con una polineuropatía sensitivo-motora axonal de predominio sensitivo. Se realizó una biopsia del nervio sural en la que se objetivó la presencia de una neuropatía axonal grave, sin depósito de amiloide ni signos compatibles con vasculitis.
El cuadro clínico del paciente fue empeorando de forma progresiva, tanto desde el punto de vista motor como sensitivo. En agosto de 2015, un tío del paciente fue diagnosticado mediante biopsia del nervio sural de una neuropatía amiloidea. Ante este dato, se solicitó un estudio de secuenciación del gen de la TTR en el que se objetivó la presencia de la mutación c.148G>A (p.Val50Met) en heterocigosis en dicho gen. Se realizó un estudio de extensión detectándose depósitos de amiloide a nivel septal y disfunción diastólica leve en el ecocardiograma. En la exploración oftalmológica no se detectaron depósitos de amiloide. Se inició tratamiento con triflusal y bisoprolol.
Se procedió a realizar un estudio genético en la familia para buscar posibles portadores de la mutación. En dicho estudio se detectó que una tía y una hermana del paciente, de 87 y 68 años de edad respectivamente, eran portadoras de la mutación Val50Met a pesar de no presentar ningún tipo de síntoma ni alteraciones en la exploración.
Paciente 2Varón de 77 años de origen guipuzcoano que acude a consultas de neurología refiriendo cansancio, dolor y hormigueos en los pies de 2 años de evolución que le dificultaban a la hora de caminar, junto con hormigueo en los dedos de las manos. No refería síntomas de disautonomía.
Como antecedentes personales, presentaba hipertensión arterial en tratamiento con indapamida y estreñimiento desde hace años en tratamiento con Plantaben. Sus padres fallecieron con 96 y 92 años sin patología neurológica. Es el pequeño de 4 hermanos de 87, 84, 82 y 81 años respectivamente, sin antecedentes familiares de interés de tipo neurológico.
La exploración neurológica objetivó parálisis de la musculatura distal de extremidades inferiores, amiotrofia de interóseos y eminencia tenar en manos, hipoestesia táctil, dolorosa y vibratoria en «calcetines» y en dedos de las manos, arreflexia universal y una marcha levemente atáxica con estepaje bilateral.
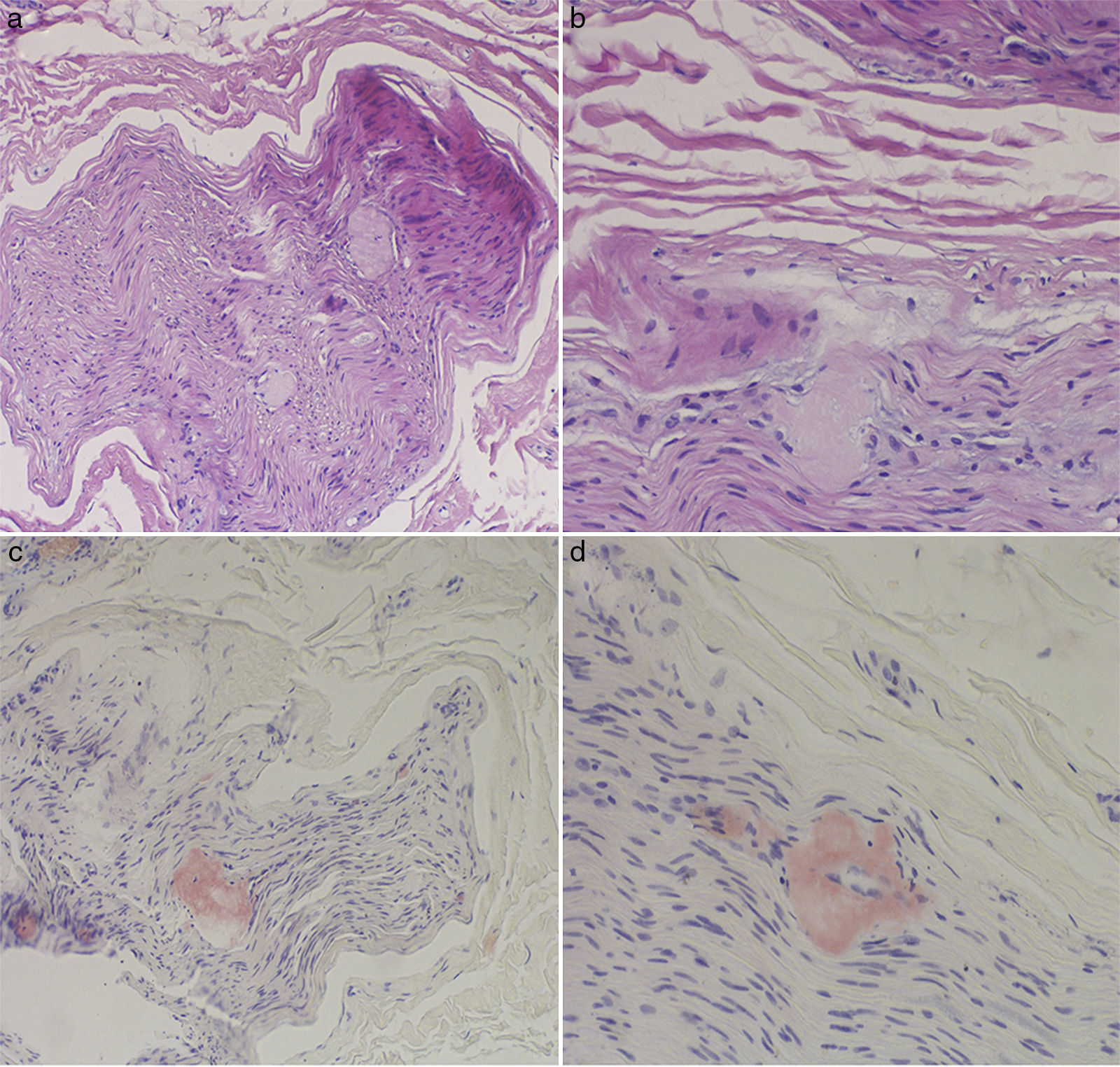
La analítica general y el sedimento de orina fueron normales. El estudio electroneurográfico fue compatible con una grave polineuropatía sensitivo-motora axonal de predominio sensitivo. Se realizó una biopsia del nervio sural en la que se detectaron depósitos de amiloide que destruían las fibras nerviosas y se teñían de rojo Congo (fig. 1).
 y tinción rojo Congo (c y d). Se objetivan depósitos amorfos hialinos eosinófilos en el endoneuro que se tiñen con rojo Congo.")
Se solicitó un estudio genético de secuenciación del gen de la TTR, objetivándose la presencia de una mutación Val50Met en heterocigosis en dicho gen.
Paciente 3Mujer de 32 años, de origen portugués, hija de un paciente fallecido con diagnóstico de PAF con mutación Val50Met en el gen de la TTR. Sus 4 tíos paternos (2 varones y 2 mujeres) así como sus 2 abuelos paternos presentaron la misma enfermedad. En 2006 acudió a la consulta de neurología para realizar un estudio genético. La exploración física y neurológica, la analítica y el estudio neurofisiológico eran normales en ese momento. El estudio genético mostró que la paciente era portadora de la misma mutación en heterocigosis. Se realizó consejo genético y diagnóstico preimplantacional ya que la paciente tenía deseos de tener un hijo biológico. En julio de 2015, acudió a revisión refiriendo dolor lumbar de gran intensidad irradiado a ambas extremidades inferiores junto con hormigueos, sensación de quemazón, alodinia y edema en los pies. Un mes antes comenzó con diarreas que alternaban con estreñimiento y distensión abdominal importante. Notaba inestabilidad y mareo al caminar. No presentaba otra clínica asociada ni signos o síntomas de hipotensión ortostática. Estaba en tratamiento con venlafaxina, alprazolam y mirtazapina por ansiedad reactiva al diagnóstico presintomático de esta enfermedad. La sensibilidad táctil, dolorosa y vibratoria, los reflejos osteotendinosos, así como el balance muscular en la exploración eran normales. El ecocardiograma mostró dilatación de la aurícula izquierda sin otras alteraciones. En el ECG-Holter se objetivaron extrasístoles ventriculares de baja densidad. La gammagrafía ósea fue normal. Ante la sospecha de los primeros síntomas de la enfermedad, se inició tratamiento con tafamidis y control del dolor neuropático con duloxetina. Posteriormente, se asoció eslicarbazepina por continuar con intenso dolor que posteriormente se retiró asociándose tapentadol y diazepam por ineficacia. En febrero de 2016, se objetiva hipoestesia térmica en los pies y en el tercio distal de las piernas, con el resto de la exploración normal. En este momento, se inician los trámites para realizar trasplante hepático, que ha sido realizado hace 2 semanas ante la persistencia de dolor neuropático muy intenso y refractario.
Paciente 4Varón de 64 años de origen guipuzcoano, exfumador, sin otros antecedentes personales de interés, que en marzo de 2005 acude a consultas de neurología por notar calambres y alodinia en las plantas de los pies, así como dificultad para subir las escaleras desde hacía 3 años. Una hermana, ya fallecida, y un hermano menor que él sufrían un cuadro similar y estaban diagnosticados de PAF. A ambos se les realizó trasplante hepático. La exploración era normal salvo por una hipoestesia táctil y vibratoria en «calcetines» y una arreflexia aquilea con hiporreflexia rotuliana. La analítica fue normal. El estudio neurofisiológico mostró una polineuropatía sensitivo-motora de carácter axonal. Se realizó estudio genético en el que se detectó la mutación Val50Met en heterocigosis. El ECG-Holter mostró extrasístoles supraventriculares de alta densidad junto con múltiples salvas de taquicardia supraventricular. El test de mesa basculante no mostró alteraciones. Se inició tratamiento con pregabalina y amitriptilina para intentar controlar el dolor neuropático. La clínica fue progresando, asociando déficit motor en extremidades inferiores que le dificultaba la marcha. En 2007 se realizó trasplante hepático. A pesar de ello, la intensidad del dolor neuropático fue aumentado, añadiéndose al tratamiento duloxetina. Asoció estreñimiento y edemas en los pies. El cuadro neurológico se mantuvo más o menos estable, con una debilidad distal en las manos y en los pies, con dolor neuropático, siendo el paciente capaz de andar con ayuda de una muleta, pero progresó la afectación cardiaca y el paciente acabó falleciendo de insuficiencia cardiaca a los 9 años del diagnóstico.
DiscusiónLa forma de presentación clínica más frecuente, descrita por Andrade en la variante portuguesa con la mutación Val50Met, es una polineuropatía sensitivo-motora junto con una disfunción autonómica de inicio a los 30-40 años2-8,10. Al inicio afecta a las fibras amielínicas y mielínicas finas (Aδ), encargadas de la transmisión del dolor y de la temperatura2,3,5. Cuanto más largo sea un axón, tanto más expuesto se verá a sufrir el depósito de amiloide y, por tanto, a alterar su función2,5. Por este motivo la patocronia seguirá un curso ascendente, desde segmentos más distales hasta alcanzar los más proximales2,3,5,7. La evolución natural de la enfermedad conduce a la muerte a los 10-15 años del inicio de los síntomas7,9,10.
Como ha quedado demostrado con los 4 casos presentados con anterioridad, este patrón clínico típico no siempre se cumple. La enfermedad puede aparecer de forma tardía (por encima de los 50 años) o cursar con una polineuropatía motora o sensitiva pura, sin manifestaciones autonómicas2-5,10.
Esta forma de presentación es la más frecuente en zonas no endémicas. También pueden presentarse como mononeuropatías que progresan hacia una multineuritis confluyente y acaban formando un patrón polineuropático2-4,8,10. El dolor neuropático puede aparecer al inicio o durante la evolución de la enfermedad, y se caracteriza por ser de tipo quemante asociado a alodinia y por tener un ritmo circadiano con empeoramiento en las últimas horas del día2,8,10. En ocasiones, como el caso de la tía del paciente 1, la mutación no es penetrante.
Esta es una enfermedad de base genética, pero no necesariamente familiar. Se transmite de forma autosómica dominante, por lo que la presencia de antecedentes familiares ayuda en el diagnóstico, como ocurrió en 3 de nuestros 4 casos2-5,7. En la mayoría de los casos en los que la enfermedad comienza alrededor de los 30 años se encuentran antecedentes familiares. Sin embargo, los casos de inicio tardío suelen ser esporádicos11. El hecho de que la penetrancia sea incompleta y que la edad de inicio pueda ser muy tardía, de modo que algunos portadores de la mutación no lleguen a desarrollar los síntomas porque fallezcan antes, puede dar lugar a la detección de casos aparentemente esporádicos2-5. También puede ocurrir que los antecedentes familiares no se reporten por desconocimiento.
Se han detectado diferentes variantes de TTR con gran heterogeneidad genotípica en todo el mundo, especialmente en países europeos. La correlación genotipo-fenotipo y los factores que influyen en la variabilidad fenotípica y en el amplio rango de edad de presentación no se conocen con exactitud2,5. La sustitución de aminoácidos únicamente no explica completamente la variabilidad en cuanto a penetrancia, patogenia y curso clínico12. En el conocimiento de la variabilidad genética de esta enfermedad es necesaria la caracterización de haplotipos definidos por 6 polimorfismos de un solo nucleótido (SNP). En Portugal y Suecia solo el haplotipo I se relacionaba previamente con la mutación. El segundo haplotipo más frecuente, el III, se observa en británicos, franceses, italianos y japoneses12. Soares et al. han identificado 10 nuevos polimorfismos, 8 de ellos por sustitución de una única base y 2 por inserción/deleción en secuencias repetidas de dinucleótidos12. Sugieren que pueda existir un efecto modulador de la edad de inicio ejercido por un locus cercano vinculado al intervalo definido por los microsatélites D18S457 y D18S456 y asociados con el haplotipo 3-2 en el cromosoma acompañante12.
Se ha detectado un fenómeno de anticipación genética en algunas familias portuguesas, suecas y japonesas con edad de inicio temprana. Este fenómeno es mayor en caso de transmisión materna5. Se han observado fenotipos menos graves con un inicio más tardío de la enfermedad y mayor duración de la misma en pacientes con mutaciones heterocigotas como Agr104His/Val50Met o Thr119Met/Val50Met5,13. Estas mutaciones adicionales han demostrado aumentar la estabilidad de los tetrámeros de TTR, por lo que se considera que tienen un «efecto protector»5,13.
Debido a la variabilidad fenotípica y al amplio rango de edad de presentación, el diagnóstico puede ser complicado. Los estudios electrofisiológicos convencionales detectan una neuropatía de carácter axonal3,5,7. No es raro que un moderado enlentecimiento en la velocidad de conducción nerviosa se sobreinterprete y valore como una forma de polineuropatía desmielinizante crónica (CIDP), quizá por el afán de ofrecer una posibilidad terapéutica a estos enfermos. En los estadios iniciales de la variante clásica, el estudio ENG puede ser normal ya que al principio solo se afectan las fibras amielínicas y mielínicas finas2,5-7.
Confirmada la existencia de una polineuropatía, el diagnóstico etiológico puede ser elusivo si no tenemos en cuenta la existencia de variantes de inicio tardío. La biopsia tisular tiene como objetivo detectar los depósitos de amiloide. Estos presentan una birrefringencia amarillo-verdosa bajo luz polarizada tras la tinción con rojo Congo2,3,5,8 que es patognomónica de la enfermedad. Sin embargo, fiar el diagnóstico etiológico a la biopsia tiene bastantes limitaciones. Por un lado, no está claro cuál es el tejido que combina el mayor rendimiento diagnóstico con el menor riesgo de consecuencias desagradables4,6-8. La biopsia del nervio es relativamente agresiva, por lo que se han postulado otros tejidos, como la grasa subcutánea o, sobre todo, las glándulas salivares1,4-8. Además, el depósito de amiloide es parcheado, de modo que la ausencia de este en la muestra tomada no excluye el diagnóstico5,8. Finalmente, la presencia de amiloide no indica necesariamente que el depósito sea de TTR6,8. Incluso, aun cuando el estudio inmunohistoquímico determine que la naturaleza de la proteína depositada es la TTR, no diferencia entre TTR nativa o mutada3,5,6,8.
Por todo ello, creemos que el test diagnóstico de elección es la secuenciación completa del gen de la TTR1,3,5-8, dado que es un gen pequeño y, con el desarrollo actual de las técnicas de diagnóstico genético, un estudio barato, sencillo, rápido y con una elevada sensibilidad y especificidad. Además, dado que en muchos casos, especialmente en zonas no endémicas, puede presentarse como formas atípicas de inicio tardío y sin antecedentes familiares conocidos, creemos que el estudio genético de TTR debe incluirse en el protocolo diagnóstico de cualquier polineuropatía. En caso de confirmarse la enfermedad, se debe realizar un estudio genético de los familiares de riesgo y aportar el consejo genético necesario1,2,7,10.
Además, es importante hacer un estudio de extensión ya que no se debe olvidar que se trata de una enfermedad multisistémica7,8.
Desde 1990 se han ido desarrollando numerosos tratamientos, algunos de ellos en fase de estudio14,15. En la actualidad, solo están aceptados el trasplante hepático y el tafamidis.
El trasplante hepático tiene como objetivo prevenir la formación de depósitos de amiloide adicionales ya que el 95% de la TTR mutada se sintetiza en el hígado2,8. Intenta estabilizar la enfermedad, pero no corrige las lesiones ya producidas. Según los datos del Familial Amyloidotic Polyneuropathy World Transplant Registry (FAPWTR), la tasa de supervivencia tras el trasplante hepático es del 85% a los 5 años, del 67% a los 10 años y del 55% a los 20 años de la intervención8,16,17. Los mejores resultados se consiguen en la variante portuguesa Val50Met de inicio precoz2,15, con una supervivencia a los 10 años del 74%. Sin embargo, los resultados son peores en otras mutaciones15 o en casos de mutaciones de Val50Met de inicio tardío, con una supervivencia a los 10 años del 44%13,16,18. El depósito de amiloide derivado de la TTR salvaje puede continuar tras el trasplante hepático y como consecuencia de ello progresar tanto la neuropatía2,8,18 como la miocardiopatía1,2. La progresión de la amiloidosis cardiaca tras el trasplante hepático no parece que ocurra en pacientes con mutaciones Val50Met y enfermedad de inicio precoz1,19,20.
El tafamidis es un fármaco estabilizador de la TTR ya que inhibe la disociación de los tetrámeros, enlenteciendo la progresión de la enfermedad1,2,21,22. Fue evaluado en un ensayo aleatorizado, doble ciego y controlado con placebo que incluyó a pacientes con la mutación Val50Met en estadio funcional 1. A los 18 meses, la respuesta al tratamiento fue mayor en el brazo tafamidis en comparación con el placebo1,2,8,23. Con base en estos resultados, ha sido aprobado por la Agencia Española de Medicamentos y Productos para el tratamiento de la PAF por TTR con mutación Val50Met sintomática en estadio funcional 11,8. También ha sido estudiado en un estudio abierto que incluía 21 pacientes con mutaciones diferentes a la Val50Met, donde se observó que el tafamidis estabiliza el tetrámero a las 6 semanas de tratamiento con respecto a la situación basal24. No hay evidencias de eficacia en formas de inicio tardío.
Aparte de estos tratamientos, no hay que olvidar la importancia del manejo de las complicaciones multisistémicas.
ConclusiónLa PAF es una enfermedad compleja, multisistémica, con amplio rango de edad de presentación que puede presentarse de formas muy diversas. El estudio genético mediante secuenciación completa del gen TTR debe incluirse en el protocolo diagnóstico de todo paciente con polineuropatía progresiva ya que presenta una elevada sensibilidad y especificidad. Disponemos de tratamientos cuya eficacia conocemos en la variante portuguesa pero no sabemos si son también útiles en otros fenotipos, por lo que es necesario obtener más datos para el futuro.
FinanciaciónDeclaramos que no se ha recibido financiación alguna.
Conflicto de interesesDeclaramos la ausencia de conflicto de intereses.
El trabajo fue premiado como Mejor Comunicación Oral en la XXXVII Reunión de la Sociedad de Neurología del País Vasco, celebrada en el hotel Monte Igueldo de Donostia los días 26 y 27 de febrero de 2016.

