




La epilepsia ausencia juvenil (EAJ) es un tipo de epilepsia generalizada idiopática que se caracteriza por la presencia de crisis de ausencia (CA) que comienzan en la adolescencia, con un EEG típico de punta-onda generalizada, y que puede acompañarse de mioclonías y crisis tónico-clónicas generalizadas (CTCG). El pronóstico a largo plazo es incierto.
Material y métodosSeleccionamos de manera retrospectiva todos los pacientes que cumplían los criterios diagnósticos de EAJ de la ILAE 1989, analizamos las variables clínicas, el tratamiento farmacológico, el estar libre de crisis y la posibilidad de retirar el tratamiento.
ResultadosEncontramos 21 pacientes, 17 mujeres y 4 varones, el 86% presentó también CTCG y el 14% mioclonías. La edad de inicio de las CA fue de 17 años (rango: 10-44). Cuatro pacientes comenzaron con CA en la edad adulta. El seguimiento medio fue de 25 años (intervalo: 10-43). El 90% recibió tratamiento con valproato y el 62% requirió politerapia. El 43% de los pacientes están actualmente libres de crisis, aunque todos ellos en tratamiento farmacológico. Todos los intentos de retirar la medicación fracasaron, pese a largos períodos sin crisis.
ConclusionesMenos de la mitad de los pacientes con EAJ están libres de crisis. El tratamiento antiepiléptico es necesario durante toda la vida a pesar de largos períodos de remisiones.
Juvenile absence epilepsy (JAE) is a generalized form of epilepsy, characterized by absence seizures (AS) initiated in adolescence, with a typical EEG showing generalized spike-wave discharges. Apart from absences, other seizure types may be observed such as myoclonia and generalized tonic-clonic seizures (GTCS). Its long-term prognosis is uncertain.
Material and methodsWe retrospectively selected all patients who met the 1989 ILAE diagnostic criteria for JAE. We analysed clinical variables, pharmacological treatment, and seizure remission with medical treatment and seizure relapse after stopping medical treatment.
ResultsWe identified 21 patients, 17 women and 4 men, 86% of whom had suffered GTCS and 14% myoclonias. Mean age at AS onset was 17 years old (range 10-44), 4 patients debuted with AS in adulthood. Mean follow up duration was 25 years (range 10-43). Ninety per cent of the patients were treated with valproate and 62% needed polytherapy. Currently 43% have achieved seizure freedom under medical treatment. All attempts to stop treatment failed, in some cases after long periods of seizure remission.
ConclusionsLess than fifty per cent of patients with JAE achieve remission, antiepileptic treatment is mandatory during all life, despite having long periods of remission.
Las crisis de ausencia típicas (CA) se definen como episodios bruscos de alteración de la conciencia junto a descargas generalizadas de punta-onda a 3 o más Hz en el EEG1. Las CA, las mioclonías y las crisis tónico-clónicas primariamente generalizadas (CTCG), solas o en combinación, son los tipos de crisis que forman parte de las epilepsias generalizadas idiopáticas (EGI)1,2. Las EGI representan alrededor del 15 o 20% de todas las epilepsias3 y se caracterizan por: estar genéticamente determinadas, tener un inicio en la infancia o adolescencia, en pacientes con desarrollo psicomotor normal, con normalidad en las pruebas de neuroimagen y un EEG con descargas de punta-onda generalizadas2. Según la clasificación de la ILAE de 19891 destacan por su prevalencia la epilepsia con ausencias infantiles (EAI), la epilepsia mioclónica juvenil (EMJ), la epilepsia con CTCG al despertar (que posteriormente en la clasificación de la ILAE 20014 se ha denominado con el término de CTCG solamente [CTCGS]) y la epilepsia ausencia juvenil (EAJ). Recientemente se ha publicado una nueva actualización de la clasificación de la ILAE de 19895 donde la dicotomía entre “síndrome epiléptico” y “enfermedad epiléptica” ha evolucionado a “síndrome electroclínico”, “constelación epiléptica”, “epilepsia de origen estructural/anatómico” y “epilepsia de causa desconocida”, en base al mejor conocimiento etiológico y sobre todo genético de la epilepsia. Las EGI antes mencionadas están todas ellas reconocidas como síndromes electroclínicos, dada su homogeneidad clínica y características electroencefalográficas.
La EMJ y la EAI son las EGI más frecuentes, y por lo tanto, las mejor estudiadas. La prevalencia de la EMJ es del 5 al 10%3 de todas las epilepsias, con un pronóstico a largo plazo excelente si se sigue el tratamiento farmacológico adecuado, aunque con una altísima tasa de recidivas si se retira6,7. La EAI tiene una prevalencia del 1,5 al 12% según las series3, con una tasa de remisión variable, en función de los criterios diagnósticos utilizados. Según los últimos criterios de Loisseau y Panayiotopoulos8, que son más restrictivos que los que recogía la clasificación de la ILAE de 19891, la tasa de remisión puede llegar al 90%. En cambio la EAJ es una entidad menos frecuente, bastante más desconocida y probablemente infradiagnosticada. La EAJ según la clasificación de la ILAE 19891 se caracteriza por la aparición de CA en pacientes adolescentes, donde las CA son menos frecuentes y tienen una menor alteración del nivel de conciencia en comparación con la EAI. Es frecuente la presencia de CTCG, que incluso pueden aparecer más precozmente que las propias CA. Los pacientes también pueden presentar mioclonías. Afecta igual a mujeres que a hombres. Se estima que tiene una prevalencia del 0,2 al 2,4% de todas las epilepsias3, pero hay muchos interrogantes a su alrededor, ya que se no se conoce con precisión su historia natural. Pocos son los estudios que han analizado el pronóstico a largo plazo de esta entidad9–12. En estas series entre el 37 y el 62% de los pacientes están libres de crisis, pero ninguno de ellos analiza la posibilidad de retirar el tratamiento farmacológico.
El objetivo principal de nuestro trabajo es analizar el pronóstico a largo plazo de la EAJ. Estudiaremos las variables clínicas, el tratamiento farmacológico, el estar o no libre de crisis, la posibilidad de retirar el tratamiento y finalmente las variables que pueden influir en el pronóstico.
Material y métodosPacientesHemos seleccionado de forma retrospectiva todos los pacientes que cumplían los criterios diagnósticos de EAJ de la ILAE 19891 (tabla 1) visitados en el Hospital de Bellvitge y en el Hospital de Sant Boi entre el año 2005 y 2008. En nuestros centros todos los pacientes son visitados como mínimo una vez al año, a pesar de tener un buen control clínico de la epilepsia, tanto si siguen o no tratamiento farmacológico. Incluimos también aquellos pacientes que iniciaron la epilepsia en la edad adulta (aunque la ILAE define como edad de inicio la pubertad, nosotros decidimos incluir también los pacientes con edad de inicio hasta los 30 años, porque es la edad máxima en la que puede existir todavía maduración cerebral, especialmente en los lóbulos frontales).
Criterios diagnósticos de la epilepsia ausencia juvenil según la ILAE 1989
| Aparición en la pubertad. Igualdad de sexos |
| Las CA son el tipo principal de crisis, con una frecuencia menor que en la EAI, pero la alteración de conciencia no es tan severa |
| Frecuente asociación con CTCG, y estas pueden aparecer más precozmente que las propias CA |
| Puede cursar con mioclonías |
| El EEG se caracteriza por punta onda generalizada mayor o igual a 3Hz |
| Buena respuesta al tratamiento médico |
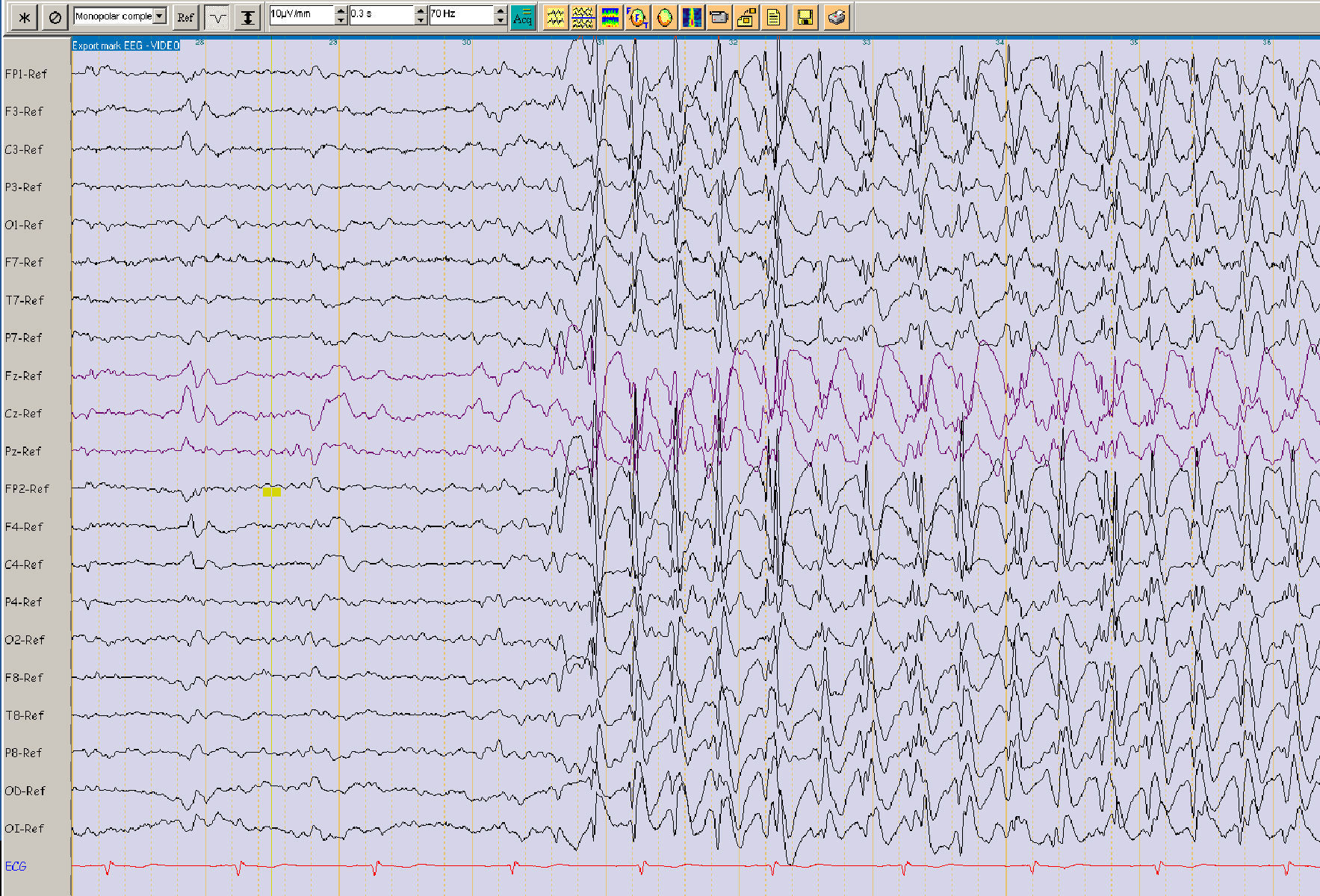
Los EEG se realizaron con colocación de los electrodos según el sistema internacional 10/20, incluyendo las activaciones de abrir/cerrar los ojos, fotoestimulación, hiperpnea y en algunos pacientes privación de sueño. Revisamos los EEG de todos los pacientes que incluimos en el estudio y todos ellos tenían descargas de punta-onda generalizada (fig. 1). En algunos casos dudosos se realizó vídeo-EEG prolongado.
Criterios de exclusión: descargas de punta-onda a 3Hz generalizadas.")
Excluimos los pacientes con antecedentes familiares de crisis febriles y epilepsia generalizada, por la posibilidad de incluir la epilepsia con crisis febriles plus, los pacientes con crisis exclusivamente fotosensibles y finalmente aquellos casos que, pese a tener una clínica altamente sugestiva, no pudimos revisar los EEG que fueron diagnósticos al inicio de la enfermedad.
VariablesVariables clínicasRecogimos en cada paciente el sexo, la edad de inicio de las CA y de las CTCG, separamos los pacientes en dos grupos: inicio pubertad (desde los 10 hasta los 17 años) y los de inicio tardío (igual o superior a 18 años), en función de la edad de la primera crisis. Consideramos necesario realizar esta separación ya que la clasificación de la ILAE 19891 únicamente reconoce como EAJ los pacientes que han iniciado las crisis en la pubertad, y no más tardíamente. También incluimos el tiempo de evolución de la enfermedad, la presencia de CTCG y si fue o no la primera manifestación, la presencia de mioclonías, la frecuencia de las CA y de las CTCG en el inicio de la enfermedad, el desarrollo de estatus epiléptico y los antecedentes familiares de epilepsia.
Variables terapéuticasIncluimos el uso de valproato (VPA), sus niveles, el número de fármacos utilizados a lo largo de la enfermedad y la necesidad de politerapia, el intento de retirada del tratamiento y en este supuesto si las crisis recidivaron.
Variables pronósticasSe recogió el estatus de libre de crisis, entendiéndose como la ausencia de crisis en los dos últimos años9,12 (en función de la percepción del paciente y la familia), con o sin tratamiento farmacológico, y también el período máximo sin crisis. Todas las variables anteriores fueron analizadas en función del estatus libre de crisis para valorar su posible relación con el pronóstico de la enfermedad. No incluimos la variable EEG en el pronóstico dado que no disponemos de los EEG evolutivos de todos los pacientes.
EstadísticaLos resultados fueron analizados con el paquete estadístico SPSS 12.0 para Windows (SPSS Inc., Chicago, IL, USA). Se realizó un análisis univariante. Las variables cualitativas fueron analizadas mediante la Chi-cuadrado unilateral (con la corrección de Yates cuando fue necesario) y para variables continuas la “t” Student y test de ANOVA. Al ser un estudio descriptivo en el que incluimos todos los pacientes con EAJ y sin criterios de exclusión, no se realizó estimación del tamaño muestral.
ResultadosDe un total de 26 pacientes que cumplían los criterios diagnósticos de la ILAE para EAJ, eliminamos un caso con antecedentes familiares de crisis febriles y epilepsia generalizada, un caso con epilepsia exclusivamente fotosensible, otro caso que comenzó a los 55 años y dos casos sin disponibilidad para revisar los EEG patológicos, obteniendo un total de 21 pacientes. Esto representa el 1,9% de todos nuestros pacientes con epilepsia.
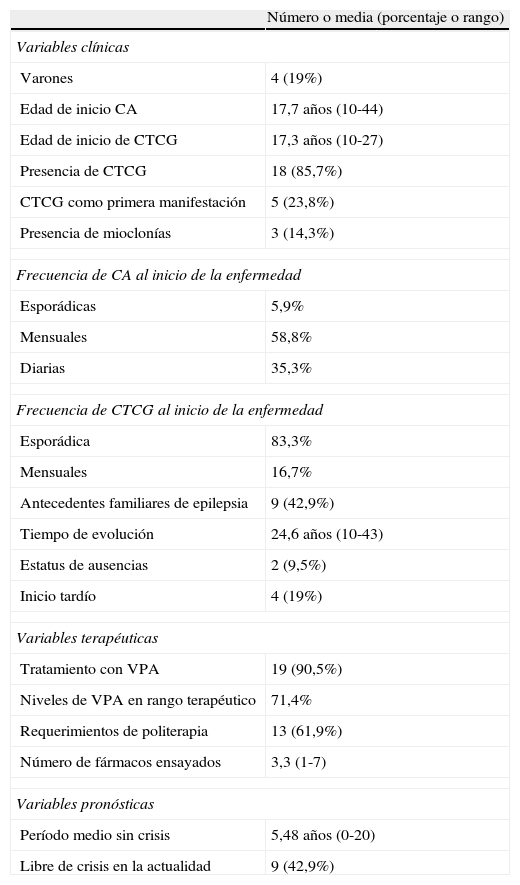
Encontramos un claro predominio del sexo femenino con 17 mujeres y 4 hombres (tabla 2). La edad media del inicio de las CA y de las CTCG fue de 17,7 y 17,3 años respectivamente (rango: 10 a 44 y de 10 a 27 años respectivamente). El 81% de los pacientes comenzaron en la pubertad, mientras que el resto fue de inicio tardío, después de los 18 años de edad. El 43% tenía antecedentes familiares de epilepsia. La gran mayoría de los pacientes presentaron CTCG en algún momento de la evolución (86%); en el 24% de los pacientes las CTCG fueron la primera manifestación clínica. El 14% del total de pacientes tenía historia de mioclonías. Las CA en el momento del diagnóstico fueron mayoritariamente mensuales (59% de los pacientes), diarias en el 35% y esporádicas en el 6%. En el 83% de los pacientes la frecuencia de las CTCG fue esporádica (menos de un episodio al año) y en el 17% fue mensual. El tiempo de evolución de la enfermedad fue muy largo, alrededor de 25 años, con un máximo de 43 y un mínimo de 10 años. Refirieron estatus de ausencias dos pacientes.
Resultados
| Número o media (porcentaje o rango) | |
| Variables clínicas | |
| Varones | 4 (19%) |
| Edad de inicio CA | 17,7 años (10-44) |
| Edad de inicio de CTCG | 17,3 años (10-27) |
| Presencia de CTCG | 18 (85,7%) |
| CTCG como primera manifestación | 5 (23,8%) |
| Presencia de mioclonías | 3 (14,3%) |
| Frecuencia de CA al inicio de la enfermedad | |
| Esporádicas | 5,9% |
| Mensuales | 58,8% |
| Diarias | 35,3% |
| Frecuencia de CTCG al inicio de la enfermedad | |
| Esporádica | 83,3% |
| Mensuales | 16,7% |
| Antecedentes familiares de epilepsia | 9 (42,9%) |
| Tiempo de evolución | 24,6 años (10-43) |
| Estatus de ausencias | 2 (9,5%) |
| Inicio tardío | 4 (19%) |
| Variables terapéuticas | |
| Tratamiento con VPA | 19 (90,5%) |
| Niveles de VPA en rango terapéutico | 71,4% |
| Requerimientos de politerapia | 13 (61,9%) |
| Número de fármacos ensayados | 3,3 (1-7) |
| Variables pronósticas | |
| Período medio sin crisis | 5,48 años (0-20) |
| Libre de crisis en la actualidad | 9 (42,9%) |
CA: crisis ausencia; CTCG: crisis tónico-clónica primariamente generalizada; VPA: valproato.
La inmensa mayoría de pacientes, el 90%, había realizado tratamiento con VPA en algún momento de la evolución, la mayoría (71%) con niveles terapéuticos. De los dos pacientes que no recibieron VPA, uno había quedado sin crisis con el primer tratamiento instaurado y en el segundo no pudimos confirmar si se había o no tratado con VPA por haber seguido controles inicialmente en otro centro. La mayoría de los pacientes libres de crisis eran tratados con VPA en monoterapia, las dosis oscilaban entre 300 y 1.500mg al día, en algunos pacientes incluso con niveles infraterapéuticos. Los pacientes libres de crisis que no recibían VPA en monoterapia eran tratados con topiramato, carbamacepina o combinación de VPA y lamotrigina. En los pacientes no libres de crisis se intentaron combinaciones de VPA con lamotrigina, topiramato, benzodiacepinas, fenobarbital, zonisamida y combinaciones entre ellos. Todos los pacientes habían recibido tratamiento farmacológico y el 62% de los casos requirió politerapia en algún momento de la enfermedad.
Se intentó retirar la medicación en 8 pacientes después de varios años sin crisis. En dos casos no se consiguió, ya que las crisis reaparecieron antes de retirar completamente la medicación. En los 6 pacientes en que se consiguió, en un caso se decidió reiniciar la medicación por presentar un EEG con muy frecuentes salvas de punta-onda generalizada sin aparente traducción clínica, y los 5 pacientes restantes volvieron a presentar crisis después de un intervalo variable de tiempo (un mes a 8 años), siendo preciso reinstaurar la medicación en todos los casos.
El 43% de los pacientes están actualmente libres de crisis, es decir, no han presentado crisis en los últimos dos años, todos ellos con tratamiento farmacológico. El período medio sin crisis durante la evolución de la enfermedad fue de 5,5 años (rango 0-20).
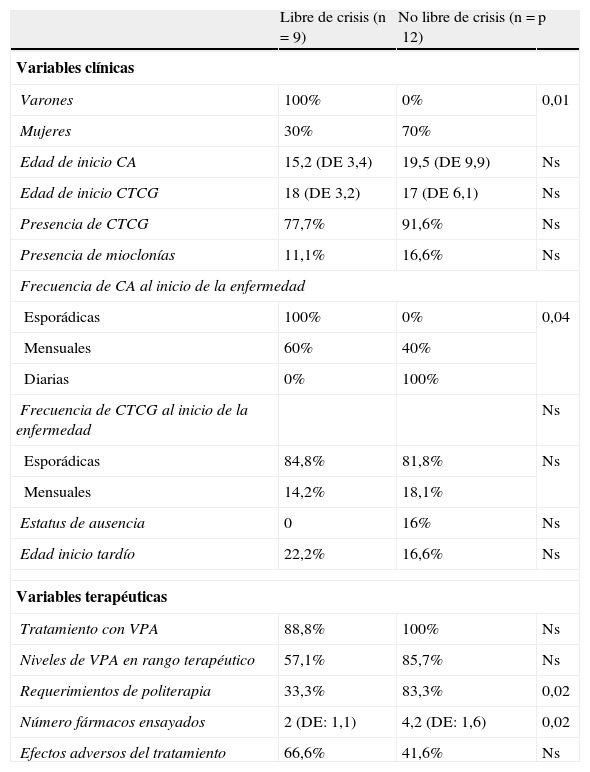
Comparamos los pacientes en función del control de las crisis y analizamos todas las variables anteriores (tabla 3). Encontramos diferencias estadísticamente significativas en función del sexo y la frecuencia de CA en el comienzo de la enfermedad. El sexo masculino tenía un mejor pronóstico que el sexo femenino, con un porcentaje libre de crisis del 100 y del 30% respectivamente (p=0,01). Los pacientes con CA diarias al inicio de la enfermedad tenían un peor pronóstico que si tenían CA mensuales, estando libres de crisis el 0 y el 60% de los pacientes respectivamente, con una p de 0,04. En relación con la medicación encontramos que los pacientes que continúan teniendo crisis tenían una mayor necesidad de politerapia y habían intentado un mayor número de fármacos (p=0,02). En relación con la presencia de CTCG, aunque el 66,6% de los pacientes sin CTCG estaban libres de crisis y tan sólo el 35,2% de los pacientes con CTCG, estos resultados no fueron estadísticamente significativos.
Variables clínicas y terapéuticas según estén o no libre de crisis
| Libre de crisis (n=9) | No libre de crisis (n=12) | p | |
| Variables clínicas | |||
| Varones | 100% | 0% | 0,01 |
| Mujeres | 30% | 70% | |
| Edad de inicio CA | 15,2 (DE 3,4) | 19,5 (DE 9,9) | Ns |
| Edad de inicio CTCG | 18 (DE 3,2) | 17 (DE 6,1) | Ns |
| Presencia de CTCG | 77,7% | 91,6% | Ns |
| Presencia de mioclonías | 11,1% | 16,6% | Ns |
| Frecuencia de CA al inicio de la enfermedad | |||
| Esporádicas | 100% | 0% | 0,04 |
| Mensuales | 60% | 40% | |
| Diarias | 0% | 100% | |
| Frecuencia de CTCG al inicio de la enfermedad | Ns | ||
| Esporádicas | 84,8% | 81,8% | Ns |
| Mensuales | 14,2% | 18,1% | |
| Estatus de ausencia | 0 | 16% | Ns |
| Edad inicio tardío | 22,2% | 16,6% | Ns |
| Variables terapéuticas | |||
| Tratamiento con VPA | 88,8% | 100% | Ns |
| Niveles de VPA en rango terapéutico | 57,1% | 85,7% | Ns |
| Requerimientos de politerapia | 33,3% | 83,3% | 0,02 |
| Número fármacos ensayados | 2 (DE: 1,1) | 4,2 (DE: 1,6) | 0,02 |
| Efectos adversos del tratamiento | 66,6% | 41,6% | Ns |
CA: crisis ausencias; CTCG: crisis tónico-clónicas generalizadas; DE: desviación estándar; Ns: no significativo.
Finalmente también comparamos los pacientes en función de la edad de inicio de la primera crisis. Los grupos fueron bastantes homogéneos, aunque los pacientes con un inicio tardío tenían menor riesgo de padecer CTCG que aquellos con un inicio precoz (33 y 92% respectivamente; p=0,04).
DiscusiónLa edad media de inicio de las CA en nuestro estudio fue de 17,7 años, siendo similar a la edad media de inicio de las CTCG, de 17,3 años; aunque es posible que la EAJ se inicie con CTCG, no es lo más frecuente1. Estos hallazgos se deben a la inclusión en nuestra muestra de 8 pacientes que comenzaron con CA en la edad adulta (4 de ellos con CTCG en la adolescencia y posterior aparición de CA en la edad adulta, y 4 pacientes sin antecedentes de crisis con un comienzo con CA en la edad adulta). Si calculamos la media de la edad de inicio de las CA y las CTCG en el resto de pacientes vemos que es de 13 y 17,9 años respectivamente, resultados superponibles al resto de estudios. Determinar la edad de inicio de las CA puede ser complicado, ya que en ocasiones pueden ser tan leves que el paciente no las identifique como patológicas. Existe por otro lado un claro predominio del sexo femenino en nuestra serie (81%). Aunque en la EAJ no se han descrito diferencias entre sexos1, sí parece existir en otras EGI como la EAI, donde se establece un predominio del sexo femenino2.
Menos de la mitad de los pacientes de nuestra muestra están libres de crisis (43%) con un seguimiento medio de 25 años. Hemos encontrado en la literatura otros estudios que analizan el pronóstico a largo plazo de la EAJ: Loiseau et al9 obtuvieron una muestra de 62 pacientes con un seguimiento muy variable (edad mínima de 20 años al terminar el estudio) y encontraron un 37% de pacientes libres de crisis; Bartolomei et al10 con 27 pacientes obtuvieron un 60% de pacientes libres de crisis con un seguimiento de unos 12 años; Tovia et al11 con una serie de 17 pacientes apuntaron a un 43,7% libres de crisis, con un seguimiento medio de 6 años, y finalmente Trinka et al12, que tienen la mayor serie con el mayor seguimiento, recogen 64 pacientes con un seguimiento medio de 25 años, obteniendo un 62% de remisiones. Bouma13 realizó un metaanálisis con 16 estudios, en el que estableció un índice de remisión muy variable, del 21 al 89% según las series, aunque incluía pacientes con EAJ y EAI. En vista de estos estudios y nuestros propios resultados, parece que la EAJ, a pesar de ser también un tipo de EGI, tiene un pronóstico peor que la EAI y la EMJ. La clasificación de la ILAE del 20014,14 postula que los pacientes con EMJ, CTCGS y EAJ deberían englobarse en el único grupo de epilepsias generalizadas de fenotipo variable de inicio en la adolescencia, aunque probablemente engloben pacientes con pronósticos distintos. Recordemos que los pacientes con EMJ que siguen un correcto tratamiento farmacológico con VPA están libres de crisis en un 90% de los casos6,7.
La mayoría de los pacientes que están libres de crisis reciben actualmente tratamiento con VPA, y al igual que en el caso de la EMJ algunos con dosis inferiores en relación con otras epilepsias focales, siendo incluso dosis infraterapéuticas7,15.
Por otro lado en nuestro estudio el 62% de los pacientes requirió politerapia en algún momento de la enfermedad. Aunque esto debe relacionarse con un mal pronóstico de la enfermedad, también podría relacionarse con un uso incorrecto de algunos fármacos antiepilépticos contraindicados en las CA, como la carbamacepina y la fenitoína. Muchos autores están de acuerdo en que el mal diagnóstico, y por lo tanto el mal tratamiento de las EGI, es la causa más frecuente de farmacorresistencia en estos tipos de epilepsias2,4,6.
La presencia de CTCG en la EAJ es muy frecuente, alrededor del 85% de los casos2, y se asocia a un peor pronóstico de la enfermedad9–12. Bouma13, en el metanálisis que realizó, encontró que el 78% de pacientes sin CTCG estaban libres de crisis, mientras que solamente el 35% de los pacientes con CTCG. Otros autores que han estudiado las CA coinciden con este hallazgo tanto en el caso de la EAI como en la EAJ8–12. En nuestra serie tan solo tenemos 3 pacientes sin CTCG, encontrando esta misma tendencia, ya que 2 de los 3 pacientes sin CTCG (66,6%) estaban libres de crisis mientras que tan solo 6 de los 17 (35,2%) pacientes con CTCG, aunque esta diferencia no era estadísticamente significativa. Por otro lado la presencia de mioclonías también se ha postulado como un factor de mal pronóstico12; nosotros no hemos encontrado diferencias en nuestra serie. De igual manera también se ha demostrado en la EMJ que los pacientes que tienen los tres tipos de crisis: CA, mioclonías y CTCG tienen más riesgo de ser farmacorresistentes16.
Según la ILAE las EGI comienzan en la infancia o en la adolescencia por definición, aunque hay numerosos autores que describen su aparición en edades más tardías17–19. Cuatro pacientes de nuestro estudio (19% del total) comenzaron de forma tardía con CA después de los 18 años, sin antecedentes de otro tipo de crisis. Estas formas tardías tendrían las mismas características clínicas que las formas clásicas, o tal vez, un mejor pronóstico18. En nuestra serie el grupo de inicio tardío tenía una menor tendencia a presentar CTCG (del 33% respecto al 92% en el grupo de inicio en la pubertad), hecho que podría correlacionarse con un mejor pronóstico. Aunque otros autores apuntan también a la existencia de síndromes que se inician exclusivamente en la edad adulta, como la EGI con ausencia fantasma20. Nuestra opinión es que los pacientes que iniciaron CA en edades tardías forman parte del mismo espectro clínico que la EAJ.
Es muy importante en los pacientes con episodios de desconexión del medio de inicio en la edad adulta, realizar siempre el diagnóstico diferencial entre las crisis focales (de origen frontal o temporal) y las CA, ya que podemos estar ante casos de EAI o EAJ no diagnosticados (sobre todo en la EAJ donde las CA pueden ser más sutiles17) o casos genuinos de CA de inicio en la edad adulta. Queremos destacar que el único paciente que no recibió tratamiento con VPA fue un paciente que comenzó en la edad adulta (19 años), diagnosticándose de epilepsia focal y siendo tratado con carbamacepina, aunque las características electroclínicas eran claramente de una EGI, inclusive una neuroimagen sin lesiones epileptógenas. Pese a todo pronóstico este paciente continúa libre de crisis con dicha medicación.
Las EGI son un diagnóstico que hay que plantearse incluso en las epilepsias de inicio en adultos. Por lo tanto la realización de un EEG, si es preciso con privación de sueño, es imprescindible para su buen diagnóstico. Encontramos una paciente que empezó a presentar CA a los 55 años, con un EEG de punta-onda generalizada a 3Hz, y con excelente respuesta al VPA; decidimos excluirla del estudio, pero tenemos la opinión de que se trata de una forma muy tardía de EAJ (aunque no estaría reconocida por la ILAE como EAJ por la edad de inicio tan tardía).
Ninguno de los estudios mencionados previamente analiza la posibilidad de retirar el tratamiento antiepiléptico en la EAJ. En nuestra serie se retiró la medicación a 6 pacientes después de un período variable sin crisis, siendo necesario reinstaurarlo en todos los casos por recidiva de crisis (excepto en un caso que fue por un EEG severamente patológico). Por lo tanto, parece que la EAJ, al igual que la EMJ, y a diferencia de la EAI, requiere mantener el tratamiento de por vida.
Las limitaciones más importantes de nuestro estudio son la reducida muestra de pacientes y sobre todo el hecho de ser retrospectivo, ya que podríamos haber perdido el espectro más benigno de la enfermedad (pacientes que habrían abandonado el seguimiento por estar libres de crisis). También hay que mencionar que la mayoría de nuestros pacientes proceden de un centro de tercer nivel, cosa que podría originar un sesgo de selección (por la inclusión de un subgrupo de pacientes de peor pronóstico). No disponemos de EEG de control en todos los casos, especialmente en aquellos sujetos que están libres de crisis. Finalmente no todos nuestros pacientes recibieron desde el inicio de la enfermedad el tratamiento más adecuado para las EGI, pudiendo condicionar un peor pronóstico a largo plazo.
En conclusión, la EAJ es una enfermedad que persiste durante toda la vida como la EMJ y otros tipos de EGI de inicio en la adolescencia, y que por lo tanto requiere un tratamiento antiepiléptico de forma indefinida. La respuesta al tratamiento en la EAJ, aun con VPA, es mucho más modesta que en la EMJ y la EAI, siendo en muchos casos necesaria la politerapia. Todos estos resultados deberían confirmarse en estudios prospectivos de mayor tamaño, para poder tomar las decisiones más acertadas en este grupo de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
El estudio fue presentado como comunicación oral en la Reunión Anual de la SEN de 2007.

