



El síndrome de hiperinsulinismo-hiperamoniemia (HI-HA) es una causa rara de hiperinsulinismo congénito1. La asociación de hipoglucemia e hiperamoniemia en un neonato es muy sugestiva de la enfermedad. Este síndrome es un error innato del metabolismo causado por mutaciones en el gen GLUD1, gen localizado en el cromosoma 10q23.3., que codifica la enzima glutamato deshidrogenasa (GDH). El gen GLUD1 contiene 13 exones que codifican los 505 aminoácidos de la enzima GDH1. Estas mutaciones conducen en último término a una hiperactividad de la GDH1,2. Las mutaciones que causan el síndrome de HI-HA han sido detectadas en los exones 11 y 12, que codifican la región antena de la enzima GDH, y en los exones 6 y 7, implicados en la codificación del sitio de unión de la GDH a la guanosina trifosfato (GTP)3,4. Las mutaciones pueden ocurrir de novo o asociadas a un patrón de herencia autosómico dominante.
El cuadro clínico en dicha entidad está constituido por hipoglucemias recurrentes sintomáticas en la infancia que se provocan por el ayuno o por una ingesta hiperproteica. La hiperamoniemia es típicamente moderada, y a diferencia de la secundaria a trastornos del ciclo de la urea, cursa de forma asintomática (es decir, sin crisis de letargia, vómitos ni cefaleas)5. El síndrome HI-HA se asocia a epilepsia6, característicamente un síndrome epiléptico generalizado de ausencias atípicas y mioclonías7, con frecuente fotosensibilidad. No es inhabitual que la epilepsia sea refractaria a la medicación antiepiléptica. Además, el síndrome se completa con otras complicaciones neurológicas como trastornos del aprendizaje, retraso mental o distonía6,8. Presentamos un nuevo caso de síndrome HI-HA por mutación del gen GLUD1. Queremos hacer hincapié en la descripción de las características fenotípicas del síndrome epiléptico de nuestro caso con crisis de ausencias mioclónicas precipitadas por la hiperventilación, así como destacar la importancia del reconocimiento de este raro síndrome con vistas a un diagnóstico precoz y correcto que evite estudios innecesarios.
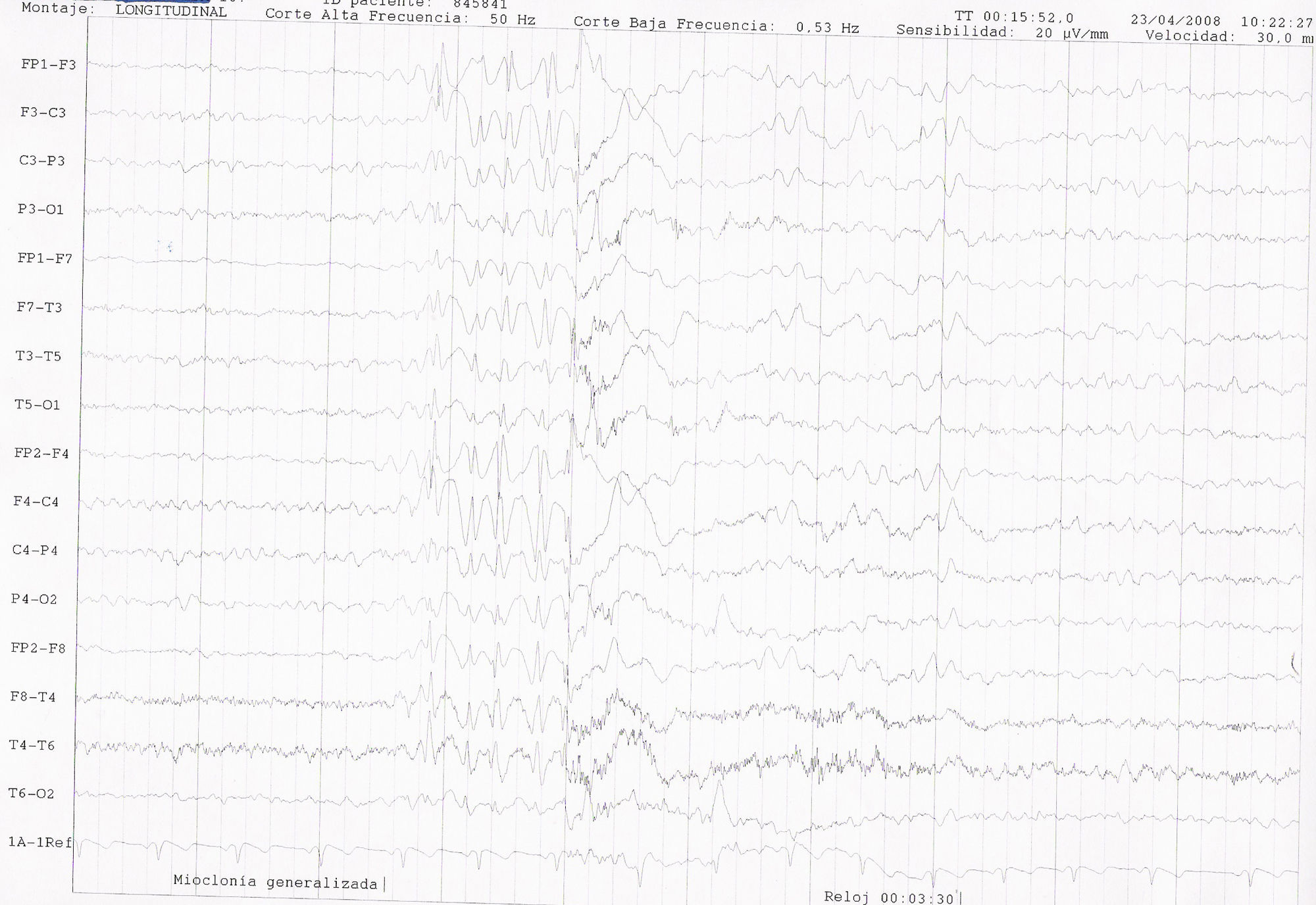
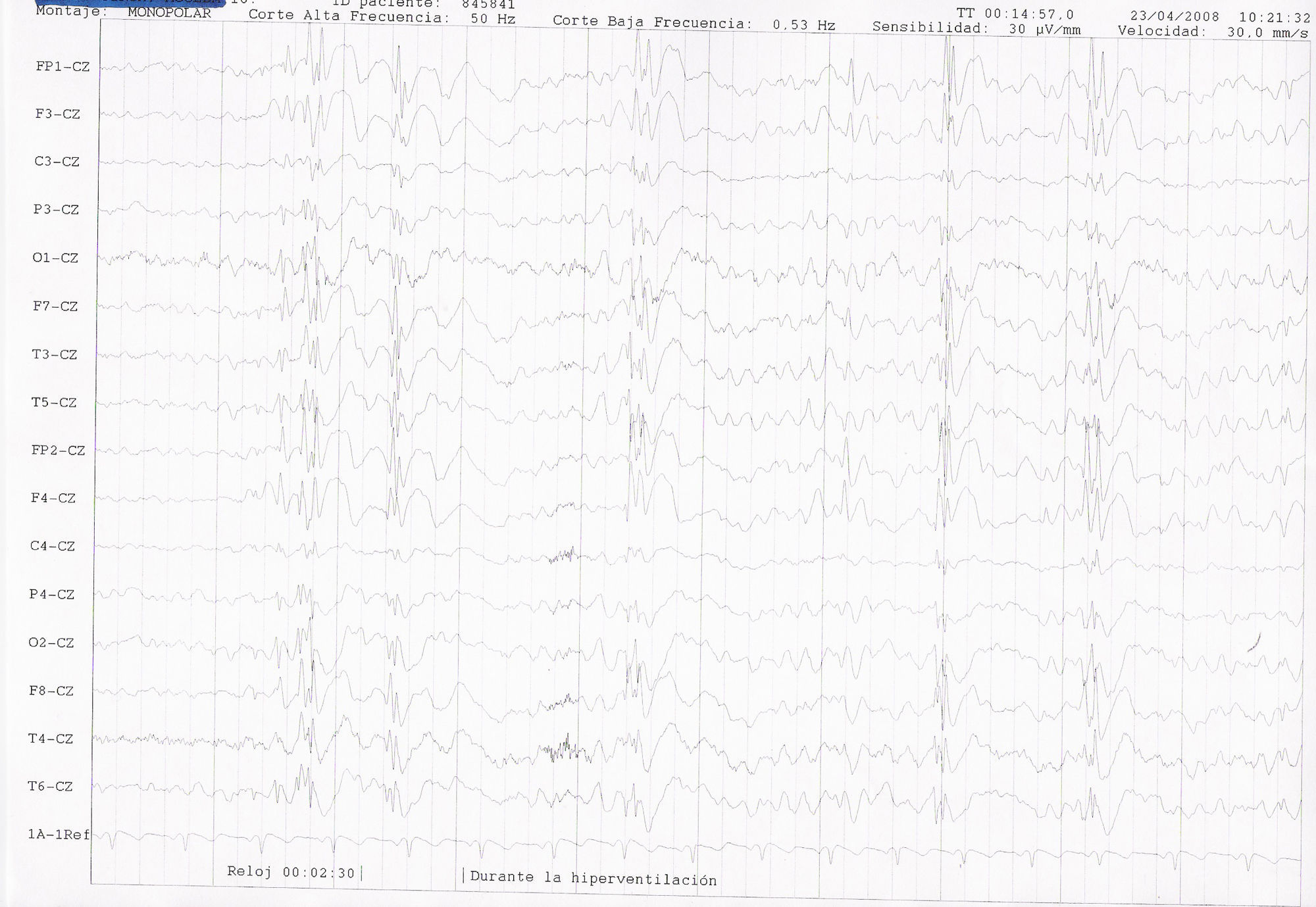
Mujer de 16 años; entre sus antecedentes familiares destaca un primo materno con epilepsia no tipificada y retraso psicomadurativo. En sus antecedentes personales: parto a término, sin complicaciones, con cesárea electiva por presentación en podálica y con peso de 3.200g, APGAR 9. Al mes de vida ingresa en la UCI por crisis de apneas severas secundarias a hipoglucemias, precisando estancia prolongada hospitalaria para su adecuado control. Se realizó, por entonces, un estudio exhaustivo etiológico, sin un diagnóstico concluyente, si bien durante el mismo se detectó la existencia de una hiperamoniemia y se instauró tratamiento para el control de la hipoglucemia con diazóxido. A la edad de 4 años comienza con crisis mioclónicas y ausencias, descritas como sacudidas erráticas y multifocales de manos, párpados, axiales y de extremidades, asociadas o no a alteración de consciencia momentánea, de varios segundos de duración, y que ocurrían de forma espontánea, tanto en vigilia como en sueño, y claramente precipitadas por la fotoestimulación y la actividad de comer. A la edad de 14 años presentó varias crisis generalizadas tonicoclónicas agrupadas en pocos días. Las crisis se han comportado durante la infancia y adolescencia como refractarias a la medicación antiepiléptica, a pesar de las distintas pautas de fármacos utilizadas (CZP, CZP+LTG, CZP+LTG+ESM, CZP+VGB, CZP+VGB+LEV, CZP+LEV, CZP+LEV+LTG, LEV+LTG+CLB). No se pautó VPA por la posibilidad de empeoramiento de la hiperamoniemia y aparición de encefalopatía asociada a la misma. Desde la edad de 16 años, y en tratamiento con la última pauta ensayada de triterapia con LEV+LTG+CLB, la paciente se encuentra razonablemente controlada de sus crisis (sin CGTC y muy ocasionales mioclonías no invalidantes). Desde el punto de vista cognitivo ha desarrollado un retraso mental borderline, con dificultades en el aprendizaje, precisando clases de apoyo, con dificultades en la integración social, baja autoestima e inestabilidad afectiva. A nivel motor destaca una leve distaxia de la marcha. En el estudio de pruebas complementarias llama la atención en la analítica una hiperamoniemia persistente con valores medios en torno a 170 micromol/l (rango 20-57). En el EEG de vigilia se aprecia una actividad fundamental ligeramente enlentecida, junto a frecuentes anomalías epileptiformes constituidas por descargas generalizadas de punta-polipunta-onda a 3,5-4Hz, breves, irregulares, de 1 a 3 segundos de duración. Este patrón se registra tanto de forma espontánea como inducido o facilitado con las maniobras de hiperventilación y con la fotoestimulación con una respuesta fotoparoxística generalizada. Dichas descargas ocasionalmente son coincidentes con mioclonías palpebrales o masivas, sin aparente afectación de consciencia (figs. 1 y 2). La RMN de cráneo no mostró alteraciones relevantes.

EEG de vigilia. Electrodos de superficie. Sistema Internacional 10-20. Sensibilidad: 15 microvolt/ mm. Velocidad de papel: 1 página/ 10 segundos. Corte alta frecuencia: 50Hz. Corte baja frecuencia: 0,53Hz. Montaje bipolar longitudinal. Se aprecia actividad epileptiforme con una descarga de punta-onda generalizada, irregular, a 3,5-4Hz, de 1,5 segundos de duración y con enlentecimiento posterior de la actividad de base, coincidente con una mioclonía generalizada.

EEG de vigilia. Electrodos de superficie. Sistema Internacional 10-20. Sensibilidad: 30 microvolt/mm. Velocidad de papel: 1 página/ 10 segundos. Corte de alta frecuencia: 50Hz. Corte de baja frecuencia: 0,53Hz. Montaje monopolar referenciado a Cz. Se registran descargas breves de polipuntas y ondas lentas hipervoltadas generalizadas durante la hiperventilación, sobre una actividad de base ligeramente enlentecida y coincidente con una crisis clínica consistente en desconexión del medio y mioclonías palpebrales y periorales.
Reevaluando el caso y ante la sospecha clínica del síndrome HI-HA se realizó recientemente el estudio molecular del gen GLUD1, detectándose una mutación en el exón 11 (p.Ser498Leu) diagnóstica de la enfermedad. Se ha realizado posteriormente el estudio genético a los padres con negatividad del mismo, tratándose de una mutación de novo.
Nuestro caso, por tanto, corresponde a un síndrome de HI-HA por mutación en el exón 11 del gen GLUD 1. El síndrome de HI-HA por mutación del gen GLUD1 es una causa rara de hiperinsulinismo congénito que se caracteriza por crisis de hipoglucemias recurrentes en la infancia e hiperamoniemia. La enfermedad se inicia en el primer año de vida en la mayoría de los casos, bien como crisis convulsiva sintomática a la hipoglucemia, bien como niño hipotónico. El control de las hipoglucemias puede retrasarse una media de 12 meses y el diagnóstico hasta 5 años6. El tratamiento de elección de las hipoglucemias es el diazóxido9 y la restricción proteica. El diazóxido actúa impidiendo el cierre de canales de K+-ATP dependiente, dificultando la despolarización de las células beta y con ello la liberación de insulina. Nuestra paciente comenzó en el periodo neonatal con crisis de apneas secundarias a hipoglucemias severas, siendo tratada desde entonces con diazóxido y precisando una estancia hospitalaria de varios meses hasta el control del cuadro. Se ha descrito el mecanismo fisiopatológico que genera el hiperinsulinismo y la hiperamoniemia1. La GDH es una enzima que media la acción reversible del glutamato a alfacetoglutarato dentro de la matriz mitocondrial. Esta acción es regulada positivamente por adenosindifosfato (ADP) y leucina y negativamente por guanosintrifosfato (GTP). En el síndrome HI-HA se incrementaría la actividad enzimática de la GDH al interferir con el efecto inhibidor de la GTP, aumentando de esta manera la producción de alfaceroglutarato, que a través del ciclo de Krebs aumentaría la producción de adenosintrifosfato (ATP). El aumento del cociente ATP/ADP provocaría un aumento de la liberación de insulina. Por otra parte, en el hígado, el incremento de la actividad de la GDH aumentaría la producción neta de amonio. La GDH es un hexámero de 6 subunidades idénticas conformada por 3 dominios10. En los pacientes con síndrome de HI-HA habría una mezcla de heterohexámeros que contendrían en igual medida subunidades mutantes y normales1. Las mutaciones más frecuentes se localizan en los exones 11 y 124. En nuestra paciente la mutación se localiza en el exón 11. También se han descrito mutaciones en los exones 6 y 711, y comunicaciones aisladas en el exón 1012. Algunos autores estiman que las mutaciones en los exones 6 y 7, que codifican el sitio de unión de la GDH a la GTP, se asocian con más frecuencia a epilepsia6. En las pocas series publicadas de casos con síndrome de HI-HA1,3,6,8,13 se ha establecido que este síndrome (además de las hipoglucemias recurrentes sintomáticas en el neonato) cursa principalmente con manifestaciones neurológicas, entre las que destaca la existencia de epilepsia en un 46-64% de los casos, el retraso mental y los trastornos de aprendizaje (51-77% según las series), siendo más raras otras manifestaciones como piramidalismo, ataxia o distonía6. El fenotipo epiléptico clásico descrito es el de una epilepsia que aparece meses después del inicio de la enfermedad, durante los primeros años de vida (si bien se han descrito casos de inicio hasta los 12 años). Las crisis epilépticas son de tipo ausencias atípicas y mioclonías palpebrales y generalizadas, aunque pueden aparecer otros tipos de crisis ya sean generalizadas (crisis generalizadas tonicoclónicas, crisis atónicas), incluso de forma mucho más rara, crisis focales. La epilepsia es refractaria a los antiepilépticos, aunque no es lo más frecuente6. Se desaconseja el uso de ácido valproico por la posibilidad de provocar resistencia a la insulina14 y empeoramiento de la amoniemia.
Nuestra paciente se comporta como un caso típico, si se nos permite la terminología, de síndrome HI-HA. Ha desarrollado una epilepsia predominantemente mioclónica, con ausencias añadidas. Las crisis se caracterizan por mioclonías tanto palpebrales, periorales, como masivas o generalizadas, de aparición espontánea, pero también facilitadas por la hiperventilación y la fotoestimulación. Como dato peculiar de nuestro caso presenta junto a la respuesta fotoparoxística una activación de las mioclonías con la hiperventilación, dato este reseñado más raramente en la literatura consultada. Durante la infancia se observó una precipitación de las crisis con la actividad de comer, circunstancia que no hemos observado en la actualidad. La epilepsia que desarrolló durante la infancia y adolescencia fue refractaria a la medicación antiepiléptica, a pesar de las distintas pautas ensayadas con fármacos a dosis óptima y eficaces tanto para crisis de ausencias como mioclónicas; posteriormente, desde los 15-16 años, presentó una mejoría en el control de las crisis, aunque actualmente precise triterapia. Pensamos que dicha mejoría está más en relación con la evolución natural de la enfermedad (mayor gravedad en la infancia y estabilización en la juventud-adultez), más que a una pauta concreta o acertada de antiepilépticos. Anotar asimismo que la paciente a pesar del uso de LTG, que teóricamente podría haber empeorado sus mioclonías, se mantiene con dicho fármaco con adecuado control de las mismas, incluso en intento de retirada ha sufrido empeoramiento de las mismas. En los últimos años se han añadido al arsenal terapéutico nuevos FAE (zonisamida, rufinamida) que pudieran tener su utilidad en este síndrome; sin embargo, no hemos encontrado revisiones al respecto.
Por otro lado no se ha demostrado una asociación cierta entre los niveles de hiperamoniemia/hipoglucemia y el daño neurológico o la epilepsia6,15. El mecanismo fisiopatológico del daño cerebral asociado al síndrome HI-HA es poco conocido, y posiblemente, complejo y multifactorial. No parece que sea debido a una lesión directa de la propia hipoglucemia. Si así fuera, esperaríamos encontrar déficits focales o epilepsia con crisis focales de forma predominante. La normalidad de la RMN craneal, sin lesiones focales, apoya esta observación. La hiperamoniemia tampoco parece desempeñar un papel primordial. En definitiva, el porqué se produce epilepsia no está claro, si bien algunos autores postulan que la mutación de la GDH produciría un defecto en el pool de glutamato, creando así un desequilibrio neurotransmisor glutamato/ GABA. Este desequilibrio podría contribuir a la epileptogénesis6. De esta forma, y en relación con la hipótesis previa, la deficiente actividad de la GDH con cúmulo de glutamato se ha implicado en el mecanismo epileptógeno de otras epilepsias, en concreto, de la epilepsia del lóbulo temporal16.
Por último, queremos resaltar la importancia de realizar un diagnóstico precoz y correcto, hoy día disponible a través del estudio genético, teniendo siempre en mente esta entidad cuando nos enfrentemos a un caso de un niño con hipoglucemia/hiperamoniemia, retraso mental y epilepsia. El síndrome HI-HA puede imitar a otras entidades que se manifiestan como epilepsias mioclónicas graves de la infancia. Debemos sospechar, por tanto, siempre esta entidad ante una epilepsia mioclónica severa de la infancia en la que aparezca el antecedente de hipoglucemia/hiperamoniemia neonatal o infantil, evitando retrasos diagnósticos y la realización de estudios innecesarios.

