



En 1996 Hinchey et al1 describieron el síndrome de leucoencefalopatía posterior reversible en relación con hipertensión arterial, alteraciones renales e inmunosupresión. Desde entonces, se han añadido numerosas entidades como potenciales desencadenantes. Conectivopatías, enfermedades hemáticas, fármacos, angiografías, transfusiones, hemodiálisis o porfiria aguda intermitente son algunos de los ejemplos2. Recientemente, se han comunicado algunos casos relacionados con la angiopatía amiloide3–7. Queremos aportar el de una paciente con este síndrome, en la que también se evidenció esta enfermedad de base.
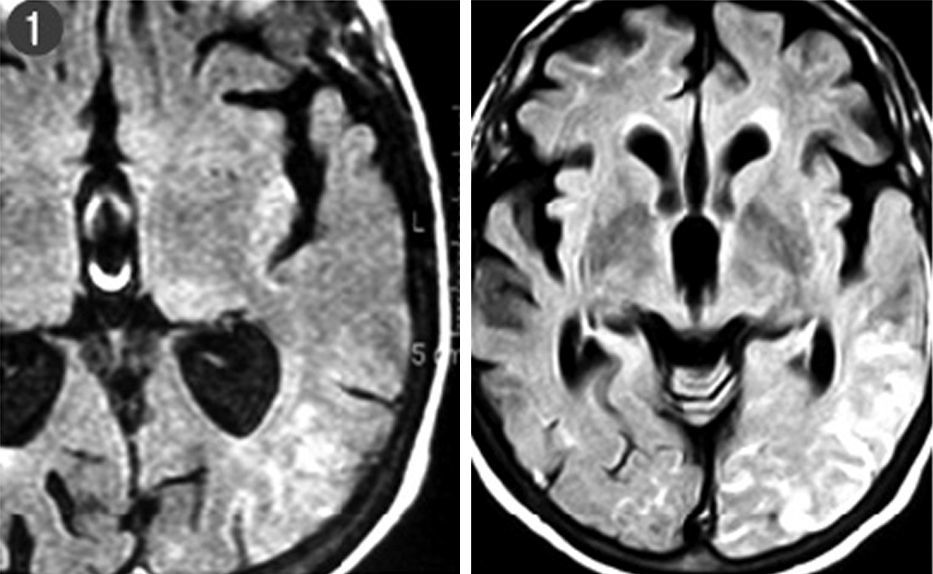
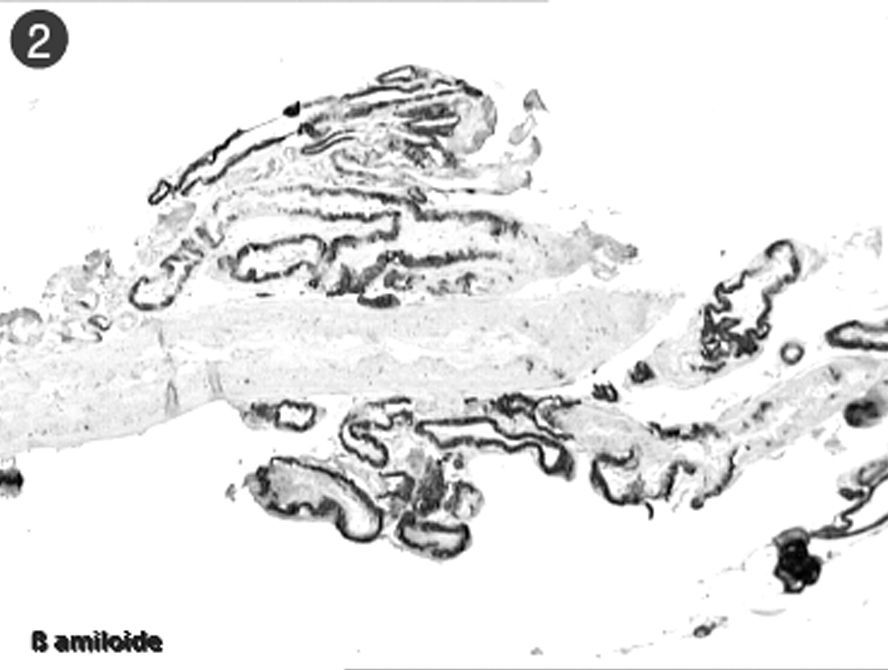
Se trata de una mujer de 73 años, hipertensa, a quien se le había realizado un trasplante renal por poliquistosis, y que estaba en tratamiento con prednisona, tacrolimus, micofenolato mofetilo y atenolol. En el estudio basal, mostró función cognitiva sin alteraciones y estado neurológico normal. En dos ocasiones, antes del trasplante, en situación de insuficiencia renal avanzada, había presentado algunos episodios de alteración del lenguaje reversibles, en el contexto de elevaciones de la presión arterial. Tres años después del trasplante, durante 18 meses, presentó cuadros bruscos, recurrentes, de incapacidad para la expresión y la comprensión del lenguaje, que remitían en aproximadamente una semana. No se acompañaban de cefalea ni de alteraciones visuales, preservaba un buen nivel de conciencia, y no se asociaban a elevaciones de la presión arterial llamativas ni a alteraciones severas de la función renal. Las manifestaciones clínicas eran similares en todos los casos. Se realizó una analítica completa, estudios de autoinmunidad, líquido cefalorraquídeo, vascular (dúplex de troncos supraórticos, Doppler transcraneal, ecocardiograma, electrocardiograma, Holter, angiografía cerebral), electroencefalograma (EEG), resonancia magnética (RM), SPECT cerebral y finalmente biopsia cerebral. En la arteriografía, se halló una placa ulcerada en la carótida interna izquierda (sintomática), que no condicionaba alteraciones hemodinámicas. En la RM craneal, además de atrofia parenquimatosa, se observó una hiperintensidad en la región parietooccipital en secuencias ponderadas en FLAIR y T2, de predominio izquierdo, con moderado realce tras la administración de contraste, y estudio normal con técnicas de difusión (fig. 1). En los EEG y vídeo-EEG, se detectaron un foco punta onda en el hemisferio izquierdo y alguna crisis eléctrica sin correlato clínico. Las demás pruebas realizadas antes de la biopsia no evidenciaron hallazgos significativos. La paciente ingresaba en el hospital reiteradamente en cada uno de estos episodios, y se fueron adoptando sucesivas medidas terapéuticas debido a la recurrencia de los cuadros. En primer lugar, recibió antiagregación, primero con aspirina y luego con clopidogrel. Posteriormente, dado el hallazgo de la placa carotídea ulcerada, que no se consideró subsidiaria de tratamiento quirúrgico, se instauró anticoagulación y tratamiento con estatinas. Por la falta de respuesta a estas terapias y los hallazgos de los EEG, se pautó tratamiento anticomicial empírico, primero con fenitoína y, más adelante, con lamotrigina. También se sustituyó el tacrolimus por everolimus8, a pesar de no encontrarse en valores tóxicos del fármaco. Con todo, la paciente seguía con alteraciones recurrentes del lenguaje (hasta seis episodios) y, a partir de los dos últimos, mostraba afasia residual. Con objeto de establecer el diagnóstico y descartar procesos infecciosos, neoplásicos e inflamatorios, se realizó una biopsia cerebral que puso de manifiesto amiloide en los vasos leptomeníngeos (fig. 2). En el parénquima cerebral, se observó una gliosis leve en la sustancia blanca, así como escasas placas difusas corticales, con ausencia de placas neuríticas y de ovillos neurofibrilares. No se detectaron cambios inflamatorios ni proliferación tumoral.


En el caso que se presenta, a pesar de que la clínica de la paciente, por su comienzo agudo, señalaba inicialmente episodios de naturaleza vascular tipo ictus, los estudios de tomografía computarizada y RM craneal nunca mostraron necrosis parenquimatosa, sino tan sólo cambios de señal inespecíficos, hiperintensos en las secuencias FLAIR y T2. La sustitución del tacrolimus se realizó debido a los potenciales efectos secundarios conocidos de este fármaco, como la encefalopatía y los trastornos del lenguaje9,10. La paciente, a pesar del trastorno del lenguaje residual, no presentó alteraciones mnésicas ni otro tipo de deterioro cognitivo adicional. La angiopatía amiloide es una vasculopatía, frecuente en la edad avanzada, que suele manifestarse en forma de hemorragias lobares. Sin embargo, en algunas ocasiones, puede hacerlo de forma exclusiva como leucoencefalopatía, bien con una distribución periventricular tipo Binswanger, más asociada a atrofia, o bien localizada en las fibras en U, inmediatamente a nivel subcortical, y en este caso suele asociar edema y tener un carácter más reversible. Por otro lado, el depósito de material amiloide tiende a ser más intenso en los lóbulos occipitales11. La explicación fisiopatológica más aceptada del síndrome de leucoencefalopatía posterior es el fallo de la capacidad de autorregulación de las arterias cerebrales, que es más prominente en regiones posteriores, por la menor densidad de inervación simpática del territorio vertebrobasilar12. En un estudio en que se compararon arterias con y sin amiloide13, se encontró que las arterias infiltradas por esta proteína presentaban adelgazamiento y degeneración de su capa media, siendo esta túnica media importante para la autorregulación. Estos hallazgos explicarían la potencial capacidad de la angiopatía amiloide para desencadenar el síndrome de leucoencefalopatía posterior. La relación entre ambas entidades ya se ha comunicado previamente en pacientes que no presentaban entre sus antecedentes otros promotores clásicos, como los que se dan en nuestro caso (hipertensión arterial, afección renal, inmunosupresores, actividad comicial). La interacción de todos ellos pudo participar en el desarrollo de los episodios y justificar la refractariedad mostrada a los cambios terapéuticos. La biopsia, extraída de la región parietooccipital posterior izquierda, aparte del depósito amiloide en los vasos, no mostró la presencia del edema parenquimatoso característico del síndrome, sino tan sólo una gliosis leve en la sustancia blanca, ya descrita en otra publicación14, posible reflejo del daño cerebral causado con el tiempo por los reiterados cuadros y en probable relación con el trastorno del lenguaje residual que apareció en los últimos meses.
La gliosis en la sustancia blanca puede estar en relación con el edema previo. Aunque la actividad comicial también puede dar lugar a gliosis, en esos casos tiende a localizarse en la corteza. En la biopsia no se observaron signos neuropatológicos de enfermedad de Alzheimer, ya que no se evidenciaron placas neuríticas ni ovillos neurofibrilares. El pequeño tamaño de la muestra no permite descartar que los hubiera en otras zonas del cerebro. La presencia de placas difusas es atribuible a la edad de la paciente. Algunos autores proponen la realización de RM craneal con secuencias de hemosiderina para tratar de filiar los casos de leucoencefalopatía de causa desconocida, en busca de microsangrados yuxtacorticales que apoyen una angiopatía amiloide, aunque éstos no necesariamente tienen que estar. También se ha planteado el tratamiento con esteroides, en los casos en que se observa en la biopsia un componente inflamatorio en relación con la angiopatía amiloide5,15, hecho que no se detectó en la muestra de nuestra paciente.
En definitiva, el síndrome de leucoencefalopatía posterior es una entidad que, además de estar causado por la encefalopatía hipertensiva, puede ser la manifestación de otras enfermedades muy diversas. Hemos de incluir entre ellas la angiopatía amiloide, para cuyo diagnóstico, en los casos en que no hay alteraciones radiológicas características de esta enfermedad, es necesario realizar una biopsia cerebral.

