



El síndrome CAPOS/CAOS (ataxia cerebelosa, arreflexia, con/sin pies cavos, atrofia óptica e hipoacusia neurosensional)1 es una rara enfermedad genética de herencia autosómica dominante, con solo 29 casos descritos en la literatura revisada a través de PubMed. Está asociado a la mutación missense en heterocigosis c.2452G>A (p.Glu818Lys) en el gen ATP1A3, y que se caracteriza por presentar episodios de descompensación neurológica asociado a encefalopatía y debilidad, que se manifiestan normalmente a una edad temprana tras un cuadro agudo de fiebre2.
Presentamos un nuevo caso de síndrome CAPOS/CAOS en el que se halla dicha variante patogénica de novo en ATP1A3 y realizamos una revisión de dicha entidad.
Mujer de 46 años, primogénita de 2 hermanos (el otro es un varón de 40 años, asintomático), de padres no consanguíneos y sin otras enfermedades de interés, que presenta en la actualidad, atrofia óptica con amaurosis bilateral, hipoacusia neurosensorial y ataxia de la marcha, que desarrolló de forma progresiva desde la infancia.
En la anamnesis detallada nos refieren que, a los 22 meses de edad, la paciente presentó 3 episodios de fiebre en el transcurso de unos 6 meses, sin enfermedad infecciosa identificada, asociando inestabilidad de la marcha parcialmente reversible entre los mismos. La biopsia muscular realizada en ese momento fue normal. Posteriormente, sobre los 5 años, desarrolla una pérdida de audición progresiva y se le realiza una biopsia de nervio sural, sin evidenciar enfermedad. En torno a los 10 años de edad, presentó un nuevo episodio de fiebre elevada sin foco infeccioso aparente, con debilidad muscular y un empeoramiento del equilibrio, que le impedía la deambulación y la realización de maniobras motoras finas. Tras dicho episodio, presentó una recuperación parcial, manteniéndose estable hasta el momento actual. Por último, sobre los 12 años apareció una disminución de la agudeza visual progresiva con atrofia óptica bilateral.
La exploración actual muestra cofosis y amaurosis bilaterales. Atrofia de ambas papilas en el fondo de ojo. Nistagmo horizontal bilateral con sacadas conservadas. Debilidad leve en orbicular de los párpados bilateral. Hipotonía leve de predominio en miembros superiores y paresia 4+/5 proximal en ambas cinturas. Arreflexia universal. Reflejo cutáneo plantar extensor derecho y tendente a la extensión el izquierdo. Leve-media dismetría en miembros superiores e inferiores. Ataxia de tronco. Marcha con aumento de la base de sustentación y Romberg positivo.
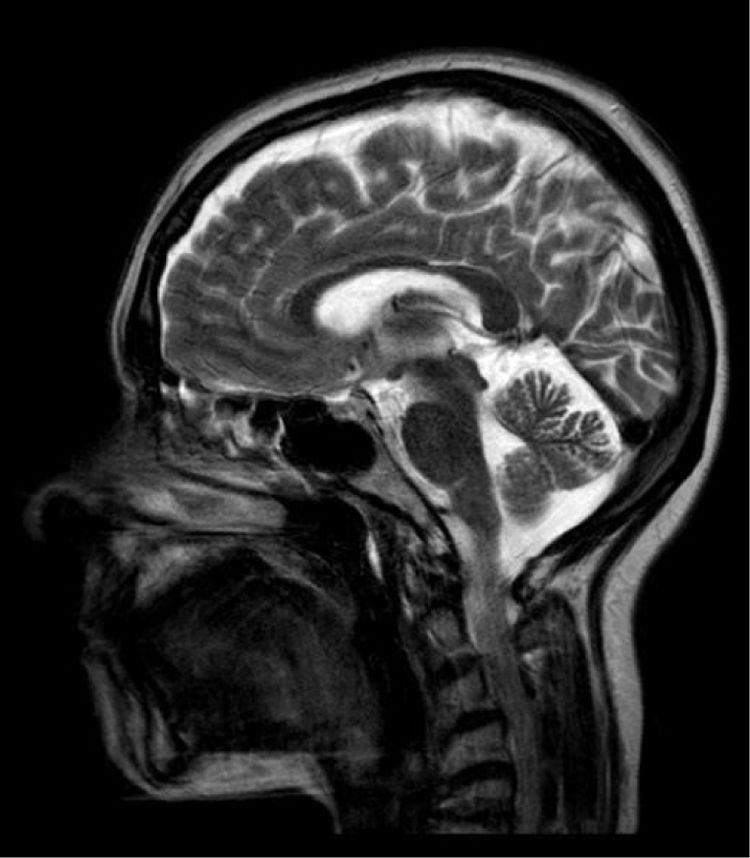
En las pruebas complementarias realizadas, destaca una resonancia magnética (RM) muscular de miembros inferiores normal, y una RM cerebral donde solo se aprecia una discreta atrofia cerebelosa mayor a la esperada para la edad de la paciente (fig. 1).

El estudio neurofisiológico, incluyendo electroneurograma y electromiograma, no arroja datos de interés.
Ante los hallazgos encontrados, solicitamos un estudio genético de miopatías mitocondriales (análisis de ADN mitocondrial más panel de genes nucleares mitocondriales) que resultó negativo. Se amplió el estudio, solicitando un panel de atrofias ópticas sindrómicas, donde se identifica la mutación en heterocigosis en el exón 18 del gen ATP1A3 (NM_152296) c.2452G>A; p.Glu818Lys, descrita como patogénica en las bases de datos consultadas (CLINVAR) y responsable del síndrome CAPOS/CAOS, con un patrón de herencia autosómica dominante2. El estudio de segregación familiar mostró que la mutación aparece de novo siendo, por tanto, un caso esporádico.
La evolución de la paciente ha sido de estabilización clínica, sin empeoramiento de los déficits presentados hasta el momento.
DiscusiónEl síndrome CAPOS/CAOS (OMIM#601338) es una entidad poco frecuente, descrita por Nicolaides et al. en 1996, y cuyo nombre refleja las características del cuadro clínico1. En 2014, Demos y van Karnebeek. asocian esta alteración con la variante heterocigota c.2452G>A (p.Glu818Lys) en el gen ATP1A33.
ATP1A3 codifica la subunidad α3 de la ATPasa Na+/K+, que se expresa principalmente en la cóclea, nervio óptico, corteza cerebelosa y las fibras nerviosas que inervan los músculos, explicando en gran medida las características clínicas de la enfermedad4.
Además del síndrome CAPOS/CAOS, entidad menos frecuente de los fenotipos clásicos, hay descritas otras entidades neurológicas que se relacionan con mutaciones en dicho gen y que muestran diferentes fenotipos clínicos como la hemiplejia alternante de la infancia (AHC; OMIM#614820) y la distonía con parkinsonismo de aparición rápida (DYT12;OMIM#128235). En los últimos 2 años se han añadido otros fenotipos como la encefalopatía epiléptica infantil temprana, la encefalopatía recurrente con ataxia cerebelosa (RECA), la ataxia de aparición rápida, las crisis autonómicas de aparición precoz y la distonía asimétrica paroxística. No obstante, el grupo de entidades clínicas relacionadas con el gen ATP1A3 seguirá expandiéndose. Algunos autores plantean la posibilidad de que se trate de un continuum fenotípico más que diferentes enfermedades alélicas5.
Típicamente, los síntomas suelen acontecer en la infancia. Los pacientes presentan episodios de ataxia inducida por cuadros febriles, evidenciándose, en algunos casos, síntomas de encefalopatía y debilidad muscular. No es raro diagnosticar en esta fase cuadros de encefalitis o presentaciones atípicas del síndrome de Guillain-Barré2. De forma habitual, existe una mejoría de la ataxia tras el episodio agudo, aunque algunos pacientes no presentan una recuperación completa. Todos los casos reportados experimentaron entre uno y 3 episodios agudos.
El signo menos constante de este cuadro es la presencia de pies cavos ya que está ausente hasta en el 70% de los casos (como en nuestra paciente), prefiriéndose según los autores la denominación de CAPOS/CAOS a la de CAPOS6. Es habitual no encontrar características miopáticas ni neuropáticas en las pruebas complementarias, situación que corroboramos en nuestro caso.
Dada la heterogeneidad clínica que caracteriza este trastorno y el grado de solapamiento que tiene con otros síndromes, es importante realizar un diagnóstico diferencial con atrofias ópticas dominantes sindrómicas, así como otras alteraciones de la cadena respiratoria mitocondrial.
No existe un tratamiento específico para esta entidad. Los autores abogan por el uso de la acetazolamida, principalmente para prevenir o reducir el déficit neurológico en los episodios febriles agudos. El mecanismo fisiopatológico para este tratamiento se sustenta en la inhibición de la anhidrasa carbónica, consiguiendo de esa forma un pH extracelular menor, lo que parece relacionarse con un mejor funcionamiento de la ATPasa Na+/K+4.

