



La enfermedad de Alzheimer (EA) es el principal trastorno neurodegenerativo que provoca una discapacidad intelectual total en los pacientes que la presentan. La elevada prevalencia a nivel mundial, así como la elevada carga socioeconómica que conlleva la EA para la sociedad en general, hace que sea considerada un importante problema de salud pública en este siglo xxi. En este trabajo se revisan los tratamientos actuales y en fase de desarrollo que actúan principalmente sobre la proteína β-amiloide.
DiscusiónLa hipótesis amiloidogénica propone que el péptido β-amiloide tiene un papel clave en esta enfermedad. Se han desarrollado varias estrategias farmacológicas diferentes con el objetivo de inhibir la formación de los péptidos β-amiloides, como son los inhibidores de β-secretasa y γ-secretasa. Además, se han desarrollado los tratamientos antiamiloide, que incluyen inmunoterapias pasivas y activas enfocadas a inhibir la agregación del péptido β-amiloide.
ConclusionesLos avances en la identificación de las bases moleculares de la EA pueden servir como modelo para comprender las causas de esta enfermedad neurodegenerativa. Sin embargo, los ensayos clínicos más recientes en 2 ensayos de fase iii con solanezumab, un anticuerpo monoclonal humanizado que promueve el aclaramiento del β-amiloide en el cerebro, indican que este anticuerpo no muestra eficacia en pacientes con EA leve, sugiriendo que hay que replantearse esta hipótesis amiloidogénica de la EA.
Alzheimer disease (AD) is a major neurodegenerative disorder which eventually results in total intellectual disability. The high global prevalence and the socioeconomic burden associated with the disease pose major challenges for public health in the 21st century. In this review we focus on both existing treatments and the therapies being developed, which principally target the β-amyloid protein.
DiscussionThe amyloidogenic hypothesis proposes that β-amyloid plays a key role in AD. Several pharmacological approaches aim to reduce the formation of β-amyloid peptides by inhibiting the β-secretase and γ-secretase enzymes. In addition, both passive and active immunotherapies have been developed for the purpose of inhibiting β-amyloid peptide aggregation.
ConclusionsProgress in identifying the molecular basis of AD may provide better models for understanding the causes of this neurodegenerative disease. The lack of efficacy of solanezumab (a humanised monoclonal antibody that promotes β-amyloid clearance in the brain), demonstrated by 2 recent Phase III clinical trials in patients with mild AD, suggests that the amyloidogenic hypothesis needs to be revised.
La enfermedad de Alzheimer (EA) es un trastorno neurodegenerativo de curso progresivo, que constituye la causa más frecuente de demencia entre la población mundial mayor de 65 años (50-70% de los casos de demencia)1. La enfermedad es actualmente crónica y progresiva, presentando déficits de múltiples funciones cerebrales (principalmente a niveles de la corteza e hipocampo), entre ellas la memoria, el pensamiento, la orientación, la comprensión, el cálculo, la capacidad de aprendizaje, el lenguaje y el juicio propio2. Las alteraciones en déficit cognitivo van acompañadas de un deterioro del control emocional y del comportamiento.
La elevada prevalencia a nivel mundial, así como, la elevada carga socioeconómica que conlleva la EA para la sociedad en general, hace que sea considerada un importante problema de salud pública. De hecho, las cifras apuntan a que será la «pandemia del siglo xxi», lo que la convierte en una enfermedad prioritaria para la investigación médica. A pesar de los grandes avances científicos y clínicos sobre la EA en los últimos 30 años, los tratamientos disponibles actualmente son solo sintomáticos, es decir, palían los síntomas de la enfermedad, actuando en diferentes niveles del proceso neuropatológico2. Aunque mejoran la calidad de vida de los pacientes, ninguno consigue realmente frenar la rápida y fatal progresión de la enfermedad.
Actualmente, hay solo 4 fármacos en el mercado aprobados para el tratamiento de la EA. Estos pertenecen a 2 grupos: inhibidores de la acetilcolinesterasa (AChEI) y los antagonistas de los receptores de ácido N-metil-D-aspártico (NMDAR). Los AChEI incluyen en este grupo el donepezilo, la rivastigmina y la galantamina2-4. El mecanismo de acción de los AChEI es aumentar la transmisión colinérgica mediante la inhibición de la acetilcolinesterasa en la hendidura sináptica y por ello podrían incrementar ligeramente la capacidad cognitiva de los pacientes con EA. La memantina es un antagonista del receptor NMDAR que reduce la excitotoxicidad por el bloqueo de este receptor inotrópico, ya que en la EA los niveles del neurotransmisor glutamato son patológicamente elevados. Ambos grupos de medicamentos están indicados para el tratamiento de pacientes con EA moderada3,4. Sin embargo, se ha demostrado que ninguno de estos fármacos aprobados representa realmente una cura para la enfermedad, ya que su efecto es solo paliativo y su eficacia disminuye con el tiempo.
Sin embargo, se están investigando nuevos tratamientos y estrategias terapéuticas con el objetivo de frenar el curso de la enfermedad, dirigidos, sobre todo, dada la complejidad neuropatológica de la EA, a múltiples dianas y pensados para ser administrados en las fases iniciales de la EA.
Para que los futuros tratamientos resulten eficaces, será necesario desarrollar nuevas técnicas diagnósticas que permitan hacer un diagnóstico precoz de la EA, en fase preclínica (antes de manifestarse los síntomas) o incluso que permitan predecir el desarrollo de esta.
La prevención de la EA es un reto realista para los investigadores; ahora bien, para hacerlo posible es necesario comprender mejor la etiología y en qué medida influyen los factores ambientales y el estilo de vida en el riesgo de desarrollar la enfermedad.
Etiología: hipótesis propuestas, factores de riesgo y protectoresRespecto a la etiología de la EA, no se conocen la causa o las causas que promueven su desarrollo, aunque se han propuesto diferentes hipótesis que ayudan a entender el complejo proceso neurodegenerativo de esta enfermedad5-8. La mayoría de los expertos coinciden en que se desarrolla como resultado de la combinación de múltiples factores de riesgo modificables y no modificables (edad, sexo, historia familiar y genética, ambientales, y además el estilo de vida) en lugar de una sola causa9-11.
Actualmente, las 2 hipótesis etiológicas propuestas más aceptadas por la comunidad científica son la hipótesis de la cascada amiloide y la de la fosforilación de la proteína tau:
- –
Hipótesis de la cascada amiloide6-9, la cual sugiere que el proceso neurodegenerativo observado en los cerebros con EA vendría dado principalmente como consecuencia de los eventos citotóxicos desencadenados por la formación, agregación y depósito de los péptidos β-amiloides (βA). Esta hipótesis ha sido muy apoyada por parte de los investigadores debido a los hallazgos genéticos en estudios de biología molecular, abriendo nuevas líneas de investigación en la búsqueda de fármacos para el tratamiento de la EA, tales como, inhibidores de la β y γ-secretasa o potenciadores de la α-secretasa4.
- –
Según esta hipótesis, el inicio de la EA seguiría el siguiente proceso: la proteína precursora del amiloide (APP) sería metabolizada por la vía amiloidogénica, lo que provocaría un exceso en la producción de péptido βA y/o un defecto de su eliminación4,5. La proteína βA se obtiene a partir del catabolismo de la APP, una proteína de la membrana plasmática con un solo dominio (una parte intracelular y otra extracelular) que se encuentra en diferentes tipos de células, entre ellas neuronas, astrocitos, oligodendrocitos y células gliales7,8. Está codificada por un gen localizado en el cromosoma 21, que al expresarse da lugar a 8 isoformas, de las cuales APP695 es la más abundante en el cerebro. Esta proteína es escindida por las enzimas α, β y γ-secretasas, y un complejo de proteínas que contienen el gen de la presenilina (PSEN1)7. En una situación fisiológica, siguiendo la vía no amiloidogénica, la APP es catabolizada por la α-secretasa, produciendo un fragmento (s)APPα que permanece en el espacio extracelular, y un fragmento carboxi-terminal de 83 aminoácidos (C83), que queda anclado en la membrana plasmática. (s)APPα regula la excitabilidad neuronal, mejora la plasticidad sináptica, el aprendizaje y la memoria, y aumenta la resistencia de las neuronas al estrés oxidativo y metabólico5-8. Sin embargo, en una situación neuropatológica, la APP se metaboliza por la vía amiloidogénica, en la que BACE (β-secretasa 1) fragmenta APP por el extremo N-terminal y la γ-secretasa lo hace por el extremo C-terminal, obteniendo los fragmentos (s)APPβ y Aβ40/42, que quedan en el espacio extracelular, y un fragmento C-terminal de 99 aminoácidos (C99), que puede ser transportado hacia el interior de la célula y translocado al núcleo, donde podría inducir la expresión de genes que promueven la muerte neuronal por apoptosis6,7.
La APP regula la supervivencia neuronal, la protección frente a estímulos externos tóxicos, el crecimiento de neuritas, la plasticidad sináptica y la adhesión celular, pero cuando se transforma en los péptidos βA 40/42 interfiere en las sinapsis, disminuye la plasticidad neuronal, altera el metabolismo energético y el de la glucosa, induce estrés oxidativo y disfunción mitocondrial, y perturba la homeostasis del calcio celular7.
- –
La escisión diferencial por la γ-secretasa produce diferentes péptidos βA: βA40 es la especie predominante, mientras que βA42 es el principal componente de las placas seniles. El péptido βA42 es a la vez más propenso a la agregación y neurotóxico que βA40 y se ha propuesto la hipótesis que representa a la especies patógenas βA. De esta manera, βA42 se oligomeriza y acumula en forma de placas seniles en el sistema límbico y la corteza asociativa, ejerciendo así efectos tóxicos en las sinapsis neuronales. En una segunda etapa, habría una respuesta glial, activación de los astrocitos y la microglía circundante, que liberaría citocinas o componentes del sistema del complemento dando lugar a respuestas inflamatorias. Asimismo, se instaura un estrés oxidativo en la neurona y se produce una alteración en la homeostasis del ion calcio, lo que provoca la hiperactivación de las proteínas cinasas y la inactivación de las fosfatasas. Por esta razón, la proteína tau se encuentra hiperfosforilada y forma los ovillos neurofibrilares, los cuales se acumulan en las sinapsis y en los cuerpos neuronales ocasionando la muerte neuronal por apoptosis y un déficit de neurotransmisores. Toda esta cascada de procesos concluye en la instauración de la demencia.
Así, tanto las proteínas βA (principalmente la β42) y tau han sido los principales objetivos para terapias modificadoras de la EA4. Desde este punto de vista, la EA podría ser prevenida o tratada eficazmente por la disminución de la producción de βA42 y la fosforilación de la proteína tau, además de la prevención de la agregación o mal plegamiento de estas proteínas, neutralizar o eliminar las formas agregadas o mal plegadas tóxicas de estas proteínas, o una combinación de estas modalidades4-9.
Asimismo, actualmente se están proponiendo hipótesis alternativas, como la de la alteración de la actividad mitocondrial, la hipótesis de la neuroinflamación y la hipótesis sobre el papel del metabolismo, concretamente el colesterol y la insulina10-14. Finalmente, se ha propuesto la hipótesis dendrítica de la EA15. Todo ello confirma la complejidad de esta enfermedad, a lo que se suma que el mecanismo de muerte neuronal por apoptosis aún no se conoce del todo.
Estrategias terapéuticas para el desarrollo de tratamientos modificadores del curso de la enfermedad de AlzheimerAnte la evidencia del incremento, en las próximas décadas, del número de casos de pacientes con EA, es necesario el desarrollo de un tratamiento que modifique el curso de la enfermedad de manera más efectiva.
Durante la última década, de 1998 a 2011, han fracasado unos 100 compuestos evaluados con el objetivo de modificar el curso de la EA, cuando ya estaban en fase de desarrollo clínico1,3. El motivo del fracaso de estos compuestos podría explicarse, como ya hemos comentado anteriormente, por la complejidad de la enfermedad debido a su etiología multifactorial y complejidad fisiopatológica. Encontrar un fármaco adecuado, y que además resulte eficaz en toda la población ensayada, es una tarea muy complicada.
Aunque todavía queden por resolver ciertos aspectos clave de la patogénesis de la EA, los avances científicos de los últimos 25 años han permitido establecer, de forma razonada, varias estrategias para el desarrollo de tratamientos con potencial para modificar el curso de la EA. Así pues, de entre las diferentes estrategias terapéuticas en las que se está trabajando, aquellas dirigidas, a reducir la formación de βA42 y la fosforilación de la proteína tau son las más importantes3. Estos 2 tipos de lesiones son las que han proporcionado los mayores avances en este campo, lo que podría ser la clave para el tratamiento de la EA en un futuro cercano.
Estrategias antiamiloideEn las 2 últimas décadas, la investigación se ha centrado principalmente en el papel del βA, siguiendo la hipótesis amiloidógena de la EA, y realizando grandes esfuerzos con el objetivo de desarrollar fármacos eficaces en el tratamiento de la EA4,6. Ahora bien, los múltiples fracasos clínicos de los compuestos en desarrollo han llevado a los investigadores a cuestionarse esta hipótesis. Sin embargo, se están investigando nuevos compuestos, junto con nuevas herramientas de diagnóstico de la EA, dado que se sospecha que la razón del fracaso podría ser la falta de biomarcadores que permitieran reclutar a los pacientes, que participan en los ensayos clínicos, antes de que llegaran a una fase muy avanzada de la enfermedad, en la que cualquier intervención terapéutica resulta inútil1.
Las diferentes estrategias anti-amiloide van dirigidas a actuar en diferentes puntos del metabolismo de la APP.
Disminución de la producción de péptidos β-A: inhibidores de las secretasasLa investigación, en el intento de disminuir la producción de βA, se ha centrado en la modulación de las vías enzimáticas encargadas del procesamiento anómalo de la APP, es decir, en la inhibición de la γ y/o β-secretasa y en la activación de la α-secretasa.
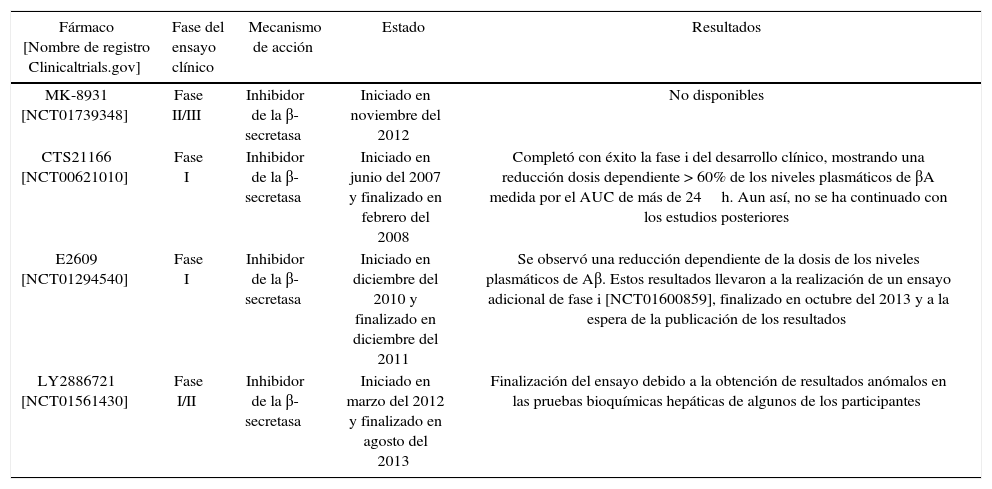
Inhibidores de la β-secretasa (BACE1)La enzima β-secretasa es la encargada de iniciar la vía amiloidogénica de procesamiento de la APP7. El desarrollo de inhibidores de esta enzima es todo un reto porque, además de la APP, la β-secretasa tiene muchos más sustratos, entre los que encontramos la neuregulina-1, implicada en la mielinización de los nervios periféricos16-18. Este hecho hace que la inhibición inespecífica de la enzima pueda dar lugar a efectos adversos16. Por otra parte, el mayor problema se encuentra en la estructura de la enzima. Al pertenecer a la clase de las aspartilproteasas, el inhibidor deberá ser una molécula grande e hidrófila, presentando problemas al tener dificultades para atravesar la barrera hematoencefálica19. Actualmente, se están investigando varios compuestos con el objetivo de superar estos obstáculos y conseguir que alguno de ellos resulte eficaz en el tratamiento de la EA19. Estudios recientes indican que 2 inhibidores de la β-secretasa, E2609 y MK-8931, son extremadamente eficaces en la reducción de la producción de los niveles de βA hasta un 80-90% en el líquido cefalorraquídeo en los seres humanos17-19.
En la tabla 1 se muestran algunos de estos compuestos que se encuentran en fase de desarrollo clínico.
Ensayos clínicos realizados con inhibidores de la β-secretasa
| Fármaco [Nombre de registro Clinicaltrials.gov] | Fase del ensayo clínico | Mecanismo de acción | Estado | Resultados |
|---|---|---|---|---|
| MK-8931 [NCT01739348] | Fase II/III | Inhibidor de la β-secretasa | Iniciado en noviembre del 2012 | No disponibles |
| CTS21166 [NCT00621010] | Fase I | Inhibidor de la β-secretasa | Iniciado en junio del 2007 y finalizado en febrero del 2008 | Completó con éxito la fase i del desarrollo clínico, mostrando una reducción dosis dependiente > 60% de los niveles plasmáticos de βA medida por el AUC de más de 24h. Aun así, no se ha continuado con los estudios posteriores |
| E2609 [NCT01294540] | Fase I | Inhibidor de la β-secretasa | Iniciado en diciembre del 2010 y finalizado en diciembre del 2011 | Se observó una reducción dependiente de la dosis de los niveles plasmáticos de Aβ. Estos resultados llevaron a la realización de un ensayo adicional de fase i [NCT01600859], finalizado en octubre del 2013 y a la espera de la publicación de los resultados |
| LY2886721 [NCT01561430] | Fase I/II | Inhibidor de la β-secretasa | Iniciado en marzo del 2012 y finalizado en agosto del 2013 | Finalización del ensayo debido a la obtención de resultados anómalos en las pruebas bioquímicas hepáticas de algunos de los participantes |
La γ-secretasa es la enzima responsable de la fase final del procesamiento de la APP por la vía amiloidogénica, dando lugar a los péptidos βA40 y βA42. Aunque la inhibición de esta enzima, en el año 2001, supuso un avance prometedor para la modificación de la enfermedad, mostrando por primera vez una disminución in vivo de la producción de β-A, el desarrollo de inhibidores de la γ-secretasa presenta problemas similares a los de los inhibidores de la β-secretasa19-21.
La actividad de la γ-secretasa procesa, además de la APP, múltiples proteínas, entre las que encontramos la proteína Notch, encargada de regular la proliferación celular, el desarrollo, la diferenciación, la comunicación y el estado de supervivencia celular20,21. Por este motivo, la inhibición inespecífica de la enzima da lugar a graves efectos adversos, los cuales plantean serias limitaciones en los ensayos clínicos.
Un ejemplo de estos fármacos desarrollados ha sido semagacestat (LY450139), un inhibidor de la γ-secretasa funcional, que se demostró que disminuía los niveles de βA en la sangre y el líquido cefalorraquídeo en los seres humanos22. El estudio clínico midió las placas de βA mediante un escáner cerebral que permite tomar imágenes de las placas amiloides en el cerebro. Los resultados de este estudio y de otros similares (NCT00762411; NCT01035138; NCT00762411) demostraron que semagacestat no disminuye la progresión lenta de la enfermedad y, además, la administración de este fármaco se asoció al empeoramiento de la cognición y la capacidad para llevar a cabo las actividades de la vida diaria22. Otro fármaco ensayado ha sido avagacestat (NCT00810147; NCT00890890; NCT00810147; NCT01079819), donde en varios ensayos clínicos se ha evaluado su farmacocinética y eficacia en la EA23-25.
Para evitar los efectos adversos derivados de estos inhibidores de la enzima γ-secretasa, se pensó en utilizar moduladores selectivos de la γ-secretasa (MSGS), los cuales bloquean la enzima alterando el procesamiento de la APP pero sin interferir con la señalización de otras vías como la Notch21. El desarrollo de los MSGS comenzó con la observación de que varios medicamentos antiinflamatorios (AINE) disminuían los niveles de péptido βA42 en células y ratones26,27. Ejemplos de estos medicamentos son ibuprofeno, sulindaco, indometacina y flurbiprofeno. R-flurbiprofeno (tarenflurbil) inhibe en mucha menor medida la ciclooxigenasa-1, ensayándose en un estudio de fase iii clínica para el tratamiento de la EA. Sin embargo, tarenflurbil e ibuprofeno fracasaron en sus respectivos ensayos clínicos27,28.
CHF5074 es un derivado antiinflamatorio no esteroideo desprovisto de actividad inhibidora de la ciclooxigenasa29. In vitro, CHF5074 se comporta como un modulador de la γ-secretasa preferentemente al inhibir la producción de βA4230,31. Como ya hemos comentado, el uso a largo plazo de AINE confiere cierta protección contra la EA, lo que llevó al estudio generalizado de los AINE frente a la producción de βA42. Sin embargo, los resultados negativos proporcionados en ensayos clínicos con AINE indican que la protección frente a la EA no es un beneficio general proporcionado por todos estos fármacos.
Un ejemplo de estos MSGS lo constituye el NIC5-15, una molécula de origen natural. Concretamente, NIC5-15 es el pinitol, un alcohol de azúcar cíclico natural32. Se encuentra en la soja y en varias otras plantas y frutas. Además, el pinitol actúa como un sensibilizador de insulina. El compuesto modula la γ-secretasa reduciendo la producción de βA, mientras que no afecta la escisión del sustrato de Notch-γ-secretasa32,33. Se ha indicado que el compuesto mejora el déficit de la función y la memoria cognitiva en modelos preclínicos de neuropatología de la EA33. Los estudios realizados en animales y ensayos en humanos han demostrado que NIC5-15 es seguro y además actúa como sensibilizador de las acciones de la insulina32. En estudios preclínicos en dosis superiores a las estudiadas previamente en los ensayos clínicos, se encontró que NIC5-15 interfiere con la acumulación de βA. Estos datos indican que NIC5-15 puede ser un agente terapéutico adecuado para el tratamiento de la EA por 2 razones: es un inhibidor de la secretasa preservando Notch y, además, es potencialmente un sensibilizador de la insulina, y se está investigando como inhibidor del proceso inflamatorio, particularmente inhibiendo la activación de la microglía.
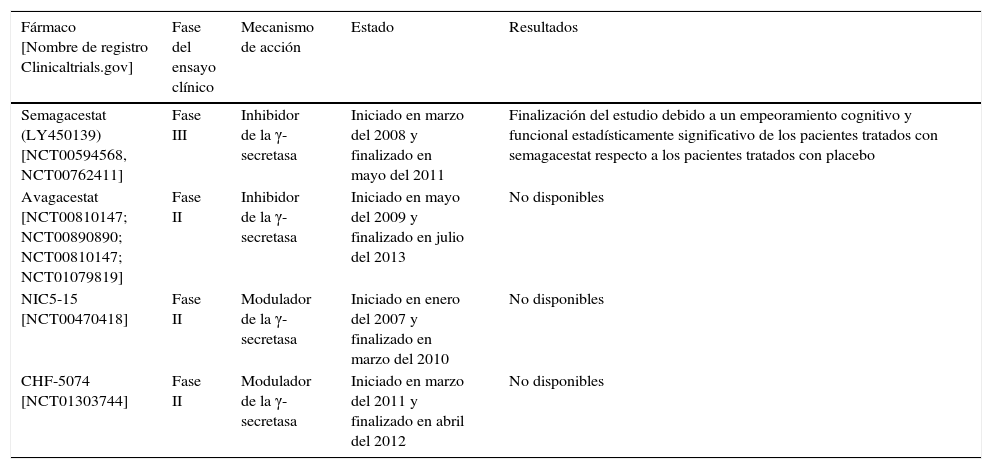
Hay varias compañías que están investigando con inhibidores/moduladores de la γ-secretasa, algunos de los cuales se muestran en la tabla 2.
Ensayos clínicos realizados con inhibidores de la γ-secretasa
| Fármaco [Nombre de registro Clinicaltrials.gov] | Fase del ensayo clínico | Mecanismo de acción | Estado | Resultados |
|---|---|---|---|---|
| Semagacestat (LY450139) [NCT00594568, NCT00762411] | Fase III | Inhibidor de la γ-secretasa | Iniciado en marzo del 2008 y finalizado en mayo del 2011 | Finalización del estudio debido a un empeoramiento cognitivo y funcional estadísticamente significativo de los pacientes tratados con semagacestat respecto a los pacientes tratados con placebo |
| Avagacestat [NCT00810147; NCT00890890; NCT00810147; NCT01079819] | Fase II | Inhibidor de la γ-secretasa | Iniciado en mayo del 2009 y finalizado en julio del 2013 | No disponibles |
| NIC5-15 [NCT00470418] | Fase II | Modulador de la γ-secretasa | Iniciado en enero del 2007 y finalizado en marzo del 2010 | No disponibles |
| CHF-5074 [NCT01303744] | Fase II | Modulador de la γ-secretasa | Iniciado en marzo del 2011 y finalizado en abril del 2012 | No disponibles |
La activación de la enzima α-secretasa conduce al procesamiento de la APP por la vía no amiloidogénica, disminuyendo por tanto la cantidad de APP disponible para la vía amiloidogénica. El resultado es la formación de un péptido βA soluble, el cual ha demostrado tener un papel neuroprotector y estimulante de la sinaptogénesis.
Así pues, la activación de la α-secretasa resulta una atractiva estrategia para el desarrollo de fármacos modificadores de la enfermedad. Se han estado investigando diferentes compuestos con potencial estimulante de la vía no amiloidogénica, entre los que encontramos agonistas de los receptores muscarínicos de la acetilcolina, glutamatérgicos, serotoninérgicos y activadores de la proteína cinasa C. Sin embargo, no se han encontrado grandes compuestos que modulen efectivamente esta vía en modelos animales, por lo que no encontramos muchas de estos compuestos en fase de ensayo clínico.
El galato de epigalocatequina (EGCG) es un flavonoide polifenólico extraído de las hojas de té verde y es considerado como el ingrediente bioactivo clave del té verde. Se ha reportado que tiene efectos beneficiosos clínicos que van desde una acción antitumoral, antiinflamatoria y neuroprotectora, y además puede tener un efecto beneficioso sobre la función cognitiva34. Se ha propuesto que el EGCG inhibe la formación de oligómeros tóxicos malformados de βA, además de activar la α-secretasa. Actualmente, se está llevando un ensayo clínico (NCT00951834) para evaluar la eficacia de EGCG en etapas tempranas de la EA.
La briostatina 1 es un modulador de la PKC y además parece ser que presenta efectos inmunomoduladores35. En animales de laboratorio se ha demostrado que incrementa la capacidad cognitiva35.
Etazolato (EHT 0202) estimula la acción neurotrófica de la α-secretasa; además, inhibe la muerte neuronal inducida por el βA, proporcionando un alivio sintomático y además modifica la progresión de la enfermedad. En un estudio clínico reciente de fase iia en 159 pacientes con EA de leve a moderada se ha demostrado que EHT0202 es seguro y generalmente bien tolerado36. Estos primeros resultados alentadores apoyan aún más el desarrollo de EHT0202 para evaluar su eficacia clínica y confirmar su tolerabilidad en una cohorte grande de pacientes con EA y durante un período más largo36.
La acitretina es un retinoide que actúa como agonista del receptor de ácido retinoico utilizado principalmente para tratar la psoriasis severa37. En modelos preclínicos que aumenta la expresión de ADAM-10, la α-secretasa de la APP humana37-39. Se ha descrito que la acitretina activa la ruta no amiloidogénica de la APP en células de neuroblastoma y reduce los niveles de βA en ratones transgénicos APP/PS137-39.
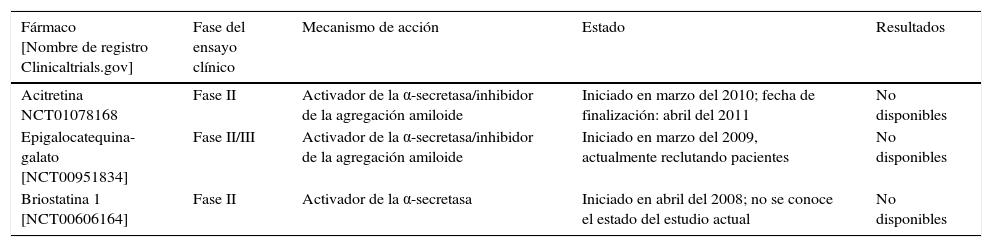
En la tabla 3 se recogen 2 de estos compuestos, el EGCG y la briostatina 1, los cuales han llegado a la fase de desarrollo clínico.
Ensayos clínicos realizados con inhibidores de la α-secretasa
| Fármaco [Nombre de registro Clinicaltrials.gov] | Fase del ensayo clínico | Mecanismo de acción | Estado | Resultados |
|---|---|---|---|---|
| Acitretina NCT01078168 | Fase II | Activador de la α-secretasa/inhibidor de la agregación amiloide | Iniciado en marzo del 2010; fecha de finalización: abril del 2011 | No disponibles |
| Epigalocatequina-galato [NCT00951834] | Fase II/III | Activador de la α-secretasa/inhibidor de la agregación amiloide | Iniciado en marzo del 2009, actualmente reclutando pacientes | No disponibles |
| Briostatina 1 [NCT00606164] | Fase II | Activador de la α-secretasa | Iniciado en abril del 2008; no se conoce el estado del estudio actual | No disponibles |
La amplia evidencia sobre la actividad neurotóxica y sinaptotóxica de los agregados amiloides constituye la base científica para el desarrollo de inhibidores de la agregación de los péptidos βA.
El único inhibidor de la agregación del βA en llegar a la fase iii es el glucosaminoglucano 3-amino ácido 1-propaneosulfonic sintético (3APS, Alzhemed, tramiprosate)40,41. Este medicamento se diseñó para interferir o antagonizar la interacción del βA con glucosaminoglucanos endógenos. Los glucosaminoglucanos se ha demostrado que promueven la agregación del βA, interfiriendo en la formación de fibrillas de amiloide y estabilizando la deposición en placas41. Sin embargo, los decepcionantes resultados del ensayo de fase iii en el año 2007 han dado lugar a la suspensión del ensayo europeo de fase iii.
La colostrinina, un complejo de polipéptido rico en prolina derivado de calostro ovino, inhibe la agregación del βA y su neurotoxicidad en ensayos celulares, y mejora el rendimiento cognitivo en modelos animales42. Aunque un ensayo de fase ii demostró ligeras mejoras en la evaluación Mini Mental State en pacientes con EA leve en un período de tratamiento de 15 meses; este efecto beneficioso no se mantuvo durante otros 15 meses de tratamiento continuado43.
El compuesto llamado escilo-inositol es capaz de estabilizar los agregados oligómeros de βA e inhibir la toxicidad del βA en el hipocampo del ratón. Se ha llevado a cabo un ensayo clínico de 18 meses en la búsqueda de dosis, seguridad y eficacia del escilo-inositol (ELND005) en los participantes con Alzheimer de leve a moderado. Se han evaluado 3 dosis de ELND005 (250, 1.000 y 2.000mg), siendo la de 250mg la más adecuada. Habrá que esperar futuros estudios clínicos a largo plazo en sujetos con EA para tener pruebas suficientes para apoyar o descartar un beneficio de ELND005 en esta enfermedad44.
Se han estado evaluando diversos compuestos que presentan un efecto antiagregante, como son el PBT1 (clioquinol) y el PBT2. Clioquinol se investigó como un tratamiento para la EA, ya que es un compuesto que bloquea la interacción entre los metales y el péptido βA en el cerebro45. Se ha propuesto que el aumento de los niveles de metales bioactivos en el envejecimiento del cerebro acelera la formación de placas amiloides, así como los procesos oxidativos neurotóxicos. La razón fundamental de la evaluación de clioquinol era que iba a evitar la acumulación de βA y, además, restauraba la homeostasis en los niveles celulares de iones como cobre y cinc. Sin embargo, estos compuestos fracasaron durante las fases ii y iii del desarrollo clínico debido a la falta de eficacia.
Compuestos que favorecen la eliminación de los agregados y depósitos amiloidesLa tercera estrategia de la ruta amiloidogénica consiste en promover el aclaramiento de los agregados y depósitos de amiloides. Para conseguirlo, se han estado evaluando 3 estrategias diferentes:
Activación de las enzimas encargadas de degradar las placas amiloidesLos agregados y las placas amiloides son degradadas por diferentes proteasas, entre las que destacan la neprilisina, enzima encargada de degradar la insulina, la plasmina, la enzima conversiva de endotelina, la enzima conversiva de angiotensina y la metaloproteinasa 946,47. En la EA, los niveles de estas enzimas disminuyen, lo que contribuye a la formación y la acumulación de placas amiloides46. A pesar de ser una atractiva estrategia antiamiloide para el desarrollo de fármacos modificadores del curso de la enfermedad, actualmente no se ha evaluado ningún activador de las proteasas debido a la carencia de especificidad de estos compuestos.
Modulación del transporte de β-amiloide desde el cerebro hacia la circulación periféricaEl transporte de βA entre el sistema nervioso central (SNC) a la circulación periférica está regulado por: 1) apolipoproteínas, donde APOE¿4 promueve el paso de βA desde la sangre hacia el cerebro; 2) la proteína relacionada a los receptores de lipoproteína de baja densidad (LRP), que incrementa el flujo de salida de βA del cerebro hacia la sangre, y 3) el receptor de los productos finales de la glucación avanzada (RAGE), que facilita la entrada de βA hacia el SNC48-51.
Aunque se hayan propuesto diferentes estrategias para incrementar el transporte de βA desde el cerebro hacia la circulación periférica, como por ejemplo la administración periférica de LRP, solo han llegado al desarrollo clínico los compuestos encaminados a inhibir/modular RAGE. Entre estos se encuentra el PF-0449470052, que fracasó en el ensayo clínico de fase ii, y el TTP4000, actualmente en ensayos clínicos de fase i (NCT01548430). El estudio finalizó en febrero del 2013 y no se han publicado resultados.
Inmunoterapia específica antiamiloideInmunoterapia activa: la inmunoterapia es la tercera estrategia enfocada a mejorar el aclaramiento de βA y la más estudiada con el objetivo de reducir la carga amiloidea en la EA. La inmunización activa (vacunación), ya sea con βA42 (la forma predominante de βA en las placas amiloides de la EA) u otros fragmentos sintéticos, se ha evaluado con éxito en modelos de ratones transgénicos de la EA. Los ensayos se basan generalmente en la estimulación de las células T, células B y la respuesta inmunitaria mediante la activación de la capacidad fagocítica de la microglía. Los resultados de los ensayos, inicialmente prometedores, han sido parcialmente suspendidos por la aparición de meningoencefalitis en algunos pacientes.
Al ensayar en pacientes la primera vacuna (AN1792), constituida por péptido βA de 42 aminoácidos, se observó que daba lugar a procesos inflamatorios neurológicos, como la meningoencefalitis aséptica, como resultado de una respuesta autoinmune anti-AN1792 mediada por células T53. Estos efectos adversos obligaron a interrumpir los ensayos clínicos de fase ii.
Los investigadores, a fin de evitar la respuesta inmunitaria no específica derivada de la inmunización con péptidos completos de βA (Aβ1-42), diseñaron unas vacunas de segunda generación, utilizando segmentos más cortos del péptido βA (Aβ1- 6), que favorecieran una respuesta humoral hacia una respuesta inmune celular.
CAD 106, diseñada por Novartis, fue la primera vacuna de segunda generación que llegó a las fases clínicas del desarrollo54. Esta ha completado recientemente la fase ii de los ensayos clínicos, en los que se observó una respuesta específica de anticuerpos βA en un 75% de los pacientes ensayados, sin dar lugar a respuestas adversas inflamatorias. La ACC-001ha completado recientemente algunos ensayos de fase ii (NCT01284387 y NCT00479557) pero, aunque hay un ensayo de fase ii en marcha (NCT01227564), la compañía farmacéutica ha desestimado continuar con la investigación. Otras vacunas, como la ACI-24, que es un es un péptido βA1-15 tetra-palmitoilado reconstituido en un liposoma, MER5101 y la AF205, se encuentran, a día de hoy, en fases preclínicas del desarrollo y se están experimentando a nivel de laboratorio55-57.
Inmunización pasiva: otro tipo de inmunoterapia bajo investigación implica la administración pasiva con anticuerpos monoclonales o policlonales dirigidos contra βA. Consiste en la administración por vía intravenosa de anticuerpos anti-βA en el paciente. De este modo, se consigue una respuesta inmunitaria anti-βA sin necesidad de una reacción proinflamatoria mediada por células T57. Los estudios en animales transgénicos han demostrado que la inmunización pasiva, además de reducir la carga amiloidogénica neuronal, mejora los déficits cognitivos, incluso antes de eliminar las placas amiloides neuronales58. Esto podría atribuirse a la neutralización de los oligómeros amiloides solubles, los cuales, se cree cada vez más, desempeñan un papel fundamental en la cascada fisiopatológica de la EA57,58.
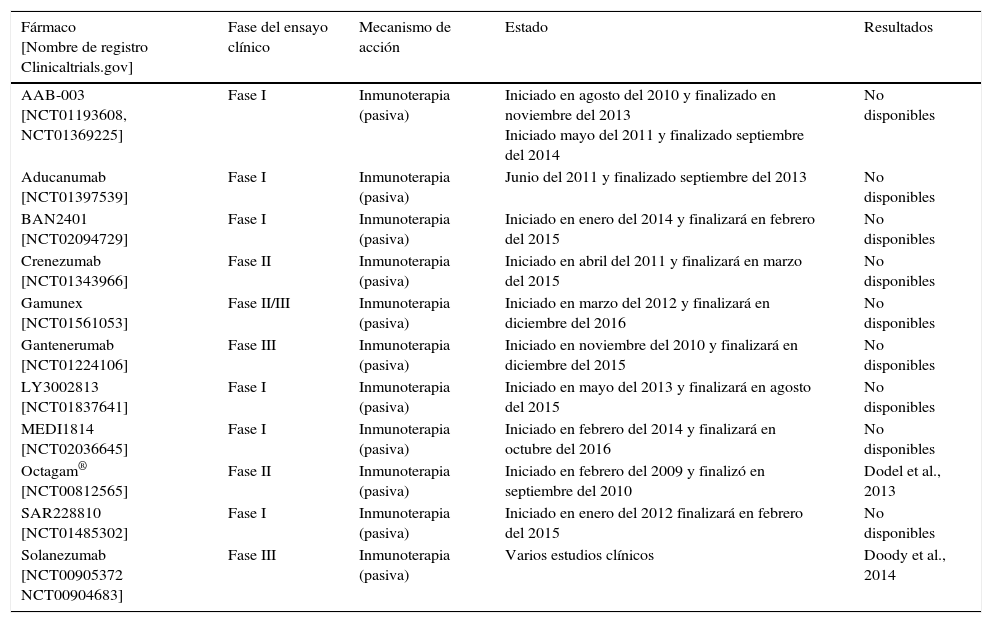
Bapineuzumab y solanezumab, los 2 anticuerpos monoclonales que han llegado a las fases más avanzadas del desarrollo clínico, fracasaron, en el año 2012, en 2 ensayos clínicos de fase iii al no mostrar los beneficios clínicos esperados en pacientes con EA leve-moderada. Bapineuzumab es un anticuerpo monoclonal humanizado contra el extremo N-terminal de la proteína βA (Aβ1-5), mientras que solanezumab es un anticuerpo monoclonal humanizado diseñado para unirse la porción central de la proteína βA (Aβ12-28)59,60. Es de destacar que bapineuzumab, a pesar de la reducción de los biomarcadores clave de EA como placa cerebral amiloide y la proteína tau fosforilada en el líquido cefalorraquídeo, falló en producir mejorías cognitivas significativas en 2 ensayos clínicos57,61. En la tabla 4 se muestran los ensayos clínicos realizados.
Ensayos clínicos realizados con inhibidores de la agregación del β-amiloide
| Fármaco [Nombre de registro Clinicaltrials.gov] | Fase del ensayo clínico | Mecanismo de acción | Estado | Resultados |
|---|---|---|---|---|
| AAB-003 [NCT01193608, NCT01369225] | Fase I | Inmunoterapia (pasiva) | Iniciado en agosto del 2010 y finalizado en noviembre del 2013 Iniciado mayo del 2011 y finalizado septiembre del 2014 | No disponibles |
| Aducanumab [NCT01397539] | Fase I | Inmunoterapia (pasiva) | Junio del 2011 y finalizado septiembre del 2013 | No disponibles |
| BAN2401 [NCT02094729] | Fase I | Inmunoterapia (pasiva) | Iniciado en enero del 2014 y finalizará en febrero del 2015 | No disponibles |
| Crenezumab [NCT01343966] | Fase II | Inmunoterapia (pasiva) | Iniciado en abril del 2011 y finalizará en marzo del 2015 | No disponibles |
| Gamunex [NCT01561053] | Fase II/III | Inmunoterapia (pasiva) | Iniciado en marzo del 2012 y finalizará en diciembre del 2016 | No disponibles |
| Gantenerumab [NCT01224106] | Fase III | Inmunoterapia (pasiva) | Iniciado en noviembre del 2010 y finalizará en diciembre del 2015 | No disponibles |
| LY3002813 [NCT01837641] | Fase I | Inmunoterapia (pasiva) | Iniciado en mayo del 2013 y finalizará en agosto del 2015 | No disponibles |
| MEDI1814 [NCT02036645] | Fase I | Inmunoterapia (pasiva) | Iniciado en febrero del 2014 y finalizará en octubre del 2016 | No disponibles |
| Octagam® [NCT00812565] | Fase II | Inmunoterapia (pasiva) | Iniciado en febrero del 2009 y finalizó en septiembre del 2010 | Dodel et al., 2013 |
| SAR228810 [NCT01485302] | Fase I | Inmunoterapia (pasiva) | Iniciado en enero del 2012 finalizará en febrero del 2015 | No disponibles |
| Solanezumab [NCT00905372 NCT00904683] | Fase III | Inmunoterapia (pasiva) | Varios estudios clínicos | Doody et al., 2014 |
Actualmente, se están realizando nuevos ensayos clínicos de fase iii con solanezumab, tanto en pacientes con enfermedad de Alzheimer (NCT01127633 y NCT01900665) como en personas mayores en fase asintomática (A4) y con riesgo elevado de perder la memoria (NCT02008357). Otro anticuerpo monoclonal, el gantenerumab, se está ensayando con el objetivo de evaluar su potencial modificador en personas con riesgo de desarrollar la EA presenil, por una mutación genética del gen DIAN-TI de carácter autosómico dominante (NCT01760005)62,63. Concretamente, en la fase iii, en enfermos con EA de leve a moderada, se les ha administrado infusiones de 400mg de solanezumab o placebo una vez al mes durante 80 semanas. Los resultados parecen indicar una tendencia a la mejora de la cognición con solanezumab en personas con EA leve, pero no parece que sea estadísticamente significativa. Por todo ello, se debe esperar a disponer de más resultados.
Paralelamente, se están realizando varios ensayos clínicos de fase iii con gantenerumab para evaluar su eficacia y seguridad en pacientes con EA leve (NCT02051608) y con EA en fase prodrómica (NCT01224106). Gantenerumab es un anticuerpo IgG1 totalmente humano diseñado para unirse con una elevada afinidad a un epítopo conformacional en las fibras de βA62,63. El fundamento terapéutico para este anticuerpo es que actúa degradando las placas amiloides mediante un proceso de reclutamiento de la microglía y activación de la fagocitosis. Los estudios experimentales en ratones transgénicos apoyan esta hipótesis64.
Crenezumab (MABT5102A) es otro anticuerpo monoclonal humanizado en fases del desarrollo clínico65. Recientemente, en abril del 2014, finalizó un ensayo clínico de fase ii en el que se evaluaba su eficacia y seguridad en pacientes con EA de leve-moderada (NCT01343966), aunque los resultados no están disponibles. Actualmente, se están realizando ensayos de fase ii con crenezumab, de los cuales, el más reciente, se inició en 2013 con el objetivo de evaluar su eficacia y seguridad en pacientes asintomáticos portadores de la mutación autosómica dominante del gen PSEN1 (NCT01998841).
Otros anticuerpos monoclonales contra βA desarrollados hasta ahora incluyen PF-04360365 (ponezumab), que se dirige a la C-terminal libre del βA y específicamente βA34-41; MABT5102A, que se une a los monómeros, oligómeros, βA y fibrillas con igualmente alta afinidad; GSK933776A, que, similar a bapineuzumab, se dirige a la secuencia N-terminal del βA65. Además, se están desarrollando otras inmunoterapias pasivas, GSK933776A, NI-101, SAR-228810 y BAN-2401, de las cuales la mayoría se encuentra en ensayos clínicos de fase i (véase la tabla 4).
Gammagard™ es una preparación de anticuerpos de plasma humano. Respecto a este preparado, se ha establecido un historial de seguridad para el uso humano en ciertas afecciones autoinmunes. Además se ha evaluado Gammagard™ en el tratamiento de la EA en un pequeño número de pacientes (NCT00818662). Estas mezclas de inmunoglobulina por vía intravenosa contienen una pequeña fracción de anticuerpos policlonales dirigidos contra el péptido βA que se cree que puede contrarrestar la toxicidad sináptica causada por βA66-68. Además, esta inmunoglobulina intravenosa tiene efectos inmunomoduladores, además de favorecer la fagocitosis de la microglía68.
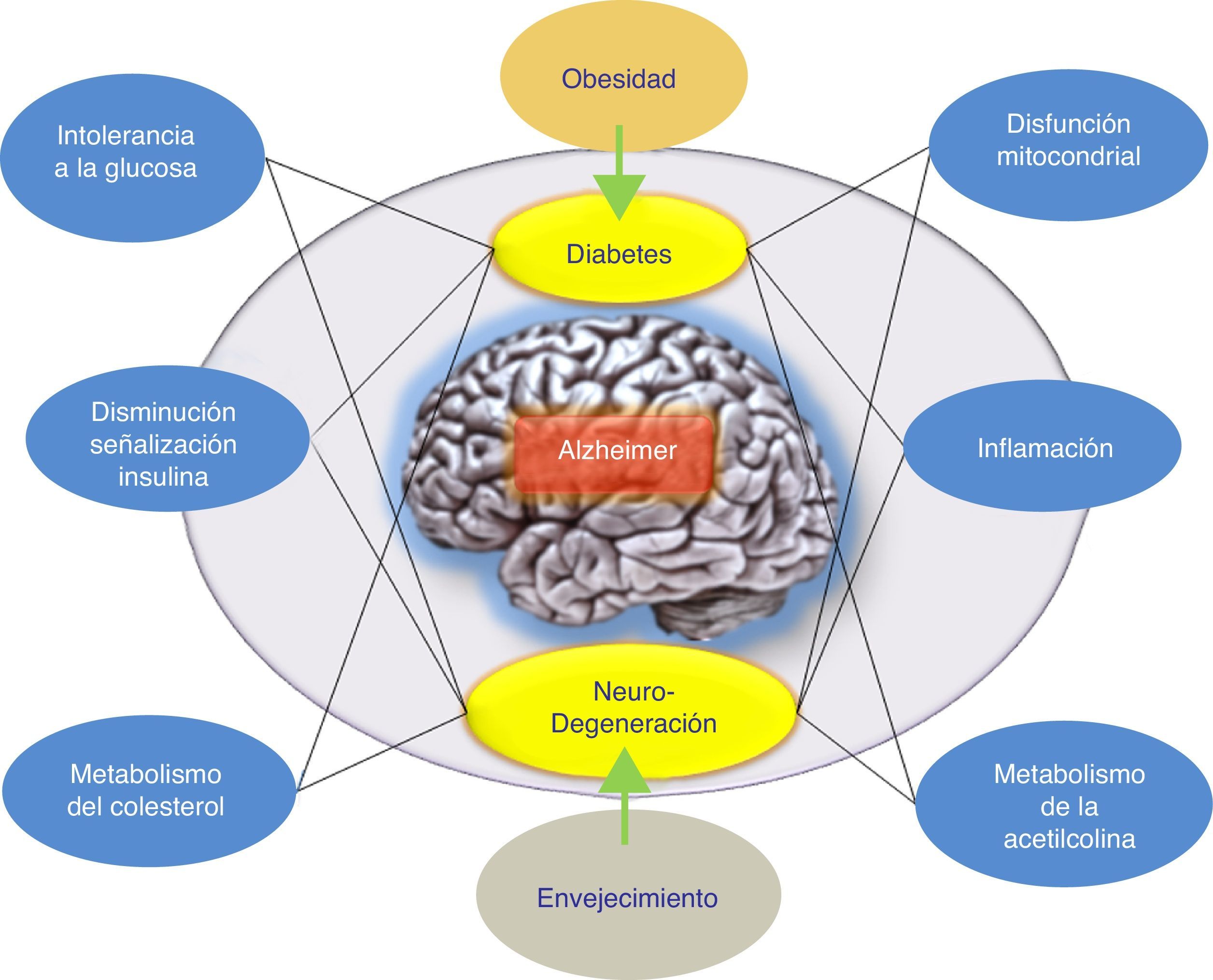
ConclusionesSe han llevado a cabo varios intentos para tratar la EA mediante la reducción de los niveles de βA cerebral. Los resultados globales en este momento han arrojado datos que indican que los fármacos antiamiloide, como grupo específico, podrían tener un efecto perjudicial sobre los síntomas de la enfermedad. Por otra parte, los resultados de los diferentes estudios realizados argumentan a favor de diferenciar cuidadosamente entre estos enfoques terapéuticos de acuerdo con el mecanismo subyacente, en lugar de agruparlos todos juntos como tratamientos antiamiloide. Además, se han propuesto hipótesis alternativas para explicar el fallo de la hipótesis amiloidogénica. Concretamente, se ha indicado la hipótesis de la respuesta adaptativa, la cual propone que el βA puede acumularse por una respuesta adaptativa frente a estímulos de estrés crónicos a nivel cerebral9. Por ello, estos estímulos de estrés constituyen las señales o vías disparadoras patogénicas del inicio tardío de la EA y, por lo tanto, serían los candidatos adecuados para la intervención terapéutica en la EA. De esta forma, el tratamiento farmacológico adecuado para frenar la EA iría a actuar sobre estos estímulos de estrés. Dichos estímulos incluirían al estrés oxidativo, la desregulación metabólica (la homeostasis del colesterol, resistencia a la insulina, etc.), los factores genéticos y la respuesta inflamatoria (fig. 1). Cada uno de estos estímulos es capaz de provocar una respuesta en la que se produciría más βA, y la naturaleza de esta respuesta determinaría la progresión clínica de la EA. Por ello, recientemente se está evaluando la insulina intranasal como una estrategia prometedora para el tratamiento de la EA, lo cual confirmaría esta hipótesis donde el péptido βA no sería el único responsable patogénico de la EA.
. El tratamiento farmacológico de la EA sería, según esta hipótesis, con fármacos que favorezcan la respuesta de la insulina. El tratamiento farmacológico adecuado para frenar la EA iría a actuar sobre estos estímulos de estrés (respuesta inflamatoria, alteración mitocondrial, etc.).")
Posible modelo propuesto para explicar el origen tardío de la enfermedad de Alzheimer. La hipótesis de la respuesta adaptativa donde se propone que el βA puede acumularse por una respuesta adaptativa frente a estímulos de estrés crónicos como desregulación metabólica (la homeostasis del colesterol o resistencia a la insulina). El tratamiento farmacológico de la EA sería, según esta hipótesis, con fármacos que favorezcan la respuesta de la insulina. El tratamiento farmacológico adecuado para frenar la EA iría a actuar sobre estos estímulos de estrés (respuesta inflamatoria, alteración mitocondrial, etc.).
Este trabajo ha sido financiado gracias a la Generalitat de Catalunya (2009/SGR00853), Ministerio de Ciencia e Innovación (SAF2011-23631), CIBERNED Instituto de Salud Carlos III, Programa PROMETEO, gobierno de Ecuador.
Conflicto de interesesLos autores declaran no tener conflicto de intereses.

