




Las distrofias musculares son miopatías genéticas, con importantes alteraciones cardiacas y si bien estas son consideradas factores de riesgo para fenómenos tromboembólicos, es baja la incidencia reportada de ataques cerebrovasculares (ACV) en estos pacientes.
Presentamos a un paciente de sexo masculino de 21 años de edad, con distrofia muscular de Duchenne, que presentó un ACV isquémico de probable etiología cardioembólica, por lo cual se inició tratamiento antitrombótico.
Sospechamos que la baja incidencia reportada de ACV en pacientes con distrofias musculares podría estar asociada a un subdiagnóstico de dichos eventos y a una baja sobrevida de estos pacientes.
The muscular dystrophies are genetic myopathies, with significant cardiac abnormalities, and while these are considered risk factors for thromboembolic events, is low reported incidence of strokes in these patients.
We present a male patient 21 years old with Duchenne muscular dystrophy, which presented an ischemic stroke probably cardioembolic, so antithrombotic treatment was started.
We suspect the low reported incidence of stroke in patients with muscular dystrophy may be associated with a sub-diagnosis of events such as low survival of these patients.
Las distrofias musculares son enfermedades del músculo estriado de origen genético. Dentro de ellas, el grupo de mayor prevalencia está constituido por las distrofinopatías, término que engloba la enfermedad de Duchenne y de Becker1. Se caracterizan por la alteración total o parcial de una proteína del citoesqueleto muscular denominada distrofina, codificada en el cromosoma X, con patrón de herencia recesivo1. La afección cardiaca se encuentra entre las principales causas de morbimortalidad de estos pacientes, produciendo alteraciones tanto estructurales (miocardiopatía dilatada, alteración de la contractilidad) como funcionales (insuficiencia cardiaca, trastornos de conducción)2. Si bien dichas alteraciones son factores de riesgo para fenómenos tromboembólicos, existen escasos reportes publicados en la literatura de ataques cerebrovasculares (ACV) agudos secundarios a dicho mecanismo2. En esta publicación presentamos un caso de ACV de etiología cardioembólica en un paciente con distrofinopatía.
Caso clínicoPaciente de sexo masculino de 21 años de edad, quien fue evaluado en la guardia externa de nuestro hospital por un cuadro de disartria y debilidad del hemicuerpo derecho de instalación súbita. Tiene como antecedente distrofia muscular de Duchenne (DMD), a expensas de presentar inmunomarcación para distrofina negativa (aislada fibra revertida) en su biopsia muscular. Durante la evolución de su enfermedad desarrolló compromiso miocárdico estructural y eléctrico, por lo que se encontraba en tratamiento con atenolol asociado a prednisona en días alternos. Desde el punto de vista funcional, el paciente presentaba un Rankin previo de 4, con necesidad de cuidados diarios y de ayuda para la deambulación.
En el ingreso hospitalario presentaba una presión arterial de 100/70mmHg y una frecuencia cardiaca regular de 100 latidos por minuto. En el examen neurológico se evidenció disartria moderada, hipotonía generalizada y cuadriparesia a predominio derecha, arreflexia generalizada e hipostesia braquiocrural derecha. Se decide su internación con diagnóstico probable de ACV en paciente joven con afectación cardiaca secundaria a su distrofia muscular. Poseía un National Institute of Health Stroke Scale (NIHSS) de 17. Se realizó una tomografía de encéfalo que evidenció cambios precoces de isquemia en su hemisferio cerebral izquierdo. Como hallazgo patológico en su laboratorio, presentaba una creatinfosfocinasa de 2.490 U/L. El electrocardiograma evidenciaba ritmo sinusal, frecuencia cardiaca de 100 latidos por minuto y ningún signo de isquemia aguda. Habiendo consultado fuera del período de ventana, se decidió iniciar tratamiento con aspirina (100mg/día) y disminuir la dosis de atenolol a la mitad.
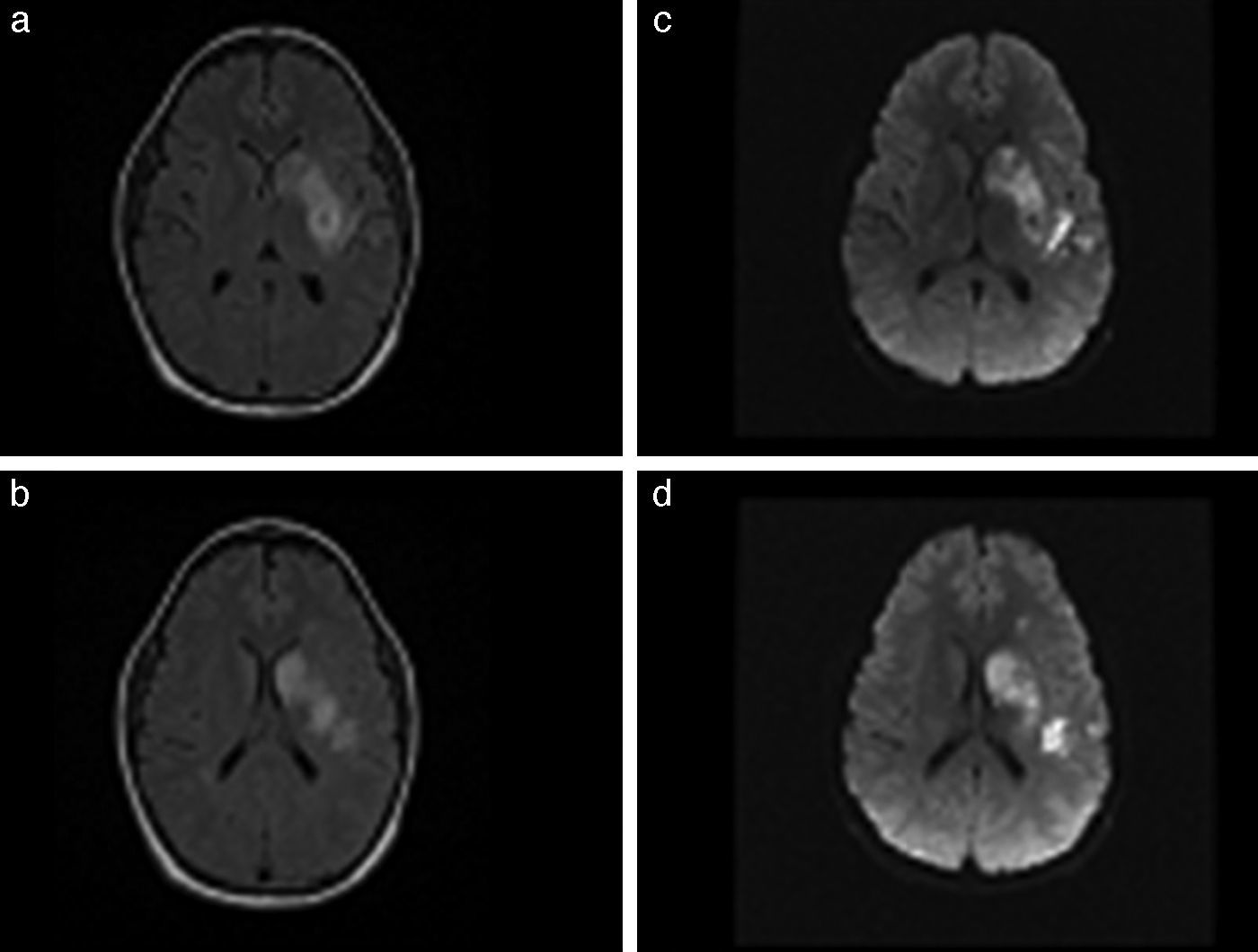
Se realizó un ecocardiograma que objetivó una función sistólica del ventrículo izquierdo moderada a severamente disminuida en forma global, con una fracción de eyección del 37% y una fracción de acortamiento del 17%, asociada a hipocinesia apical, sin evidencia de trombos intracavitarios. Se realizó Holter de 24 h que evidenció ritmo sinusal con extrasístoles ventriculares polifocales de alta densidad horaria, de distribución homogénea, asociadas a duplas con alteraciones basales de la repolarización. La resonancia magnética (RM) de encéfalo evidenció un infarto subagudo cortico/subcortical fronto-temporo-parietal izquierdo con leve transformación hemorrágica (fig. 1). La angiografía por RM de vasos intra y extracraneales con contraste no objetivó anomalías. Por lo tanto, se interpretó el cuadro como un ACV isquémico de origen cardioembólico, por lo que se inició anticoagulación con acenocumarol a los 10 días del evento, previa realización de una tomografía de cerebro control. El paciente fue dado de alta con una leve mejoría en su examen neurológico, sin presentar déficit sensitivo y con menor déficit motor derecho respecto al ingreso, con un NIHSS de 15 y un Rankin de 4.
 con restricción de señal en secuencia de difusión (c y d) en territorio de la arteria cerebral media izquierda (Ml), compatible con infarto cerebral estadio agudo/subagudo asociado a un pequeño foco de transformación hemorrágica en el brazo posterior de cápsula interna izquierda.")
RM de encéfalo. Se evidencia lesión hiperintensa en secuencia de FLAIR (a y b) con restricción de señal en secuencia de difusión (c y d) en territorio de la arteria cerebral media izquierda (Ml), compatible con infarto cerebral estadio agudo/subagudo asociado a un pequeño foco de transformación hemorrágica en el brazo posterior de cápsula interna izquierda.
Las distrofinopatías son las enfermedades musculares hereditarias más frecuentes, producto de mutaciones en el gen de la distrofina en el cromosoma Xp21, lo cual ocasiona un déficit total o parcial, o anomalías en la proteína distrofina, tanto en el músculo estriado esquelético como cardiaco. La DMD es la forma más grave de este grupo, la cual se caracteriza por mutaciones, principalmente deleciones, que llevan a la ausencia total de la distrofina3. La incidencia es de 1/3.500 varones nacidos vivos, manifestándose inicialmente por dificultad para la marcha y caídas frecuentes a la edad 3-4 años. Conforme progresa la enfermedad, pierden la capacidad de deambular, requiriendo utilización de silla de ruedas durante la adolescencia4.
El compromiso cardiaco en la DMD comienza precozmente, alrededor de los 6-7 años, aumentando la incidencia de miocardiopatía dilatada con la edad4. La cardiopatía está presente en forma clínica o subclínica en el 90% de los casos, siendo la insuficiencia cardiaca la causa de muerte en el 20% de los pacientes5. El grado de afectación cardiaca varía de acuerdo con el estadio de la enfermedad y el tipo de mutación5. Se reconocen 2 patrones de compromiso cardiaco: en etapas tempranas de la enfermedad se producen alteraciones en la motilidad del ventrículo izquierdo, mientras que la cardiomiopatía dilatada es más frecuente en la segunda década de la vida6. Se manifiesta inicialmente con alteraciones electrocardiográficas del ritmo y la conducción, evidenciándose luego anormalidades en el ecocardiograma, como alteraciones focales de la motilidad, disfunción ventricular e insuficiencia cardiaca por depresión de la función sistólica en etapas tardías de la enfermedad5. El tratamiento de la insuficiencia cardiaca congestiva con inhibidores de la enzima convertidora de angiotensina, diuréticos y betabloqueantes mejora la condición clínica de estos pacientes2.
Las alteraciones cardiacas relacionadas con la DMD son capaces de provocar ACV isquémicos por favorecer el desarrollo de fenómenos tromboembólicos, Sin embargo, en lo reportado por el trabajo de Hanajima y Kawai se observó en un total de 665 con DMD una incidencia del 0,75% de ACV7 y Biller et al. en su estudio en un total de 52 pacientes no encontraron eventos cerebrovasculares8. Los reporte de casos en la literatura indexada sobre ACV en pacientes con DMD son un total de 5, en todos los casos los pacientes presentaban una disfunción sistólica del ventrículo izquierdo moderada a severa, por el que se explicaba en fenómeno cardioembólico del evento cerebrovascular2,9-11. Esta complicación de tan baja incidencia reportada podría deberse una tasa importante de subdiagnóstico, que probablemente se deba a múltiples factores, entre ellas la presencia de debilidad muscular generalizada en estos pacientes, que podría enmascarar el desarrollo de déficit motor de origen cerebrovascular o la presentación frecuente de atrofia muscular severa, la cual podría dificultar la aparición de signos piramidales2. Otro factor probable asociado a la baja incidencia de cardioembolias cerebrales podría ser la baja sobrevida de un grupo importante de pacientes con esta patología, los cuales, a pesar de presentar cardiopatías de alto riesgo de embolia, no llegan a desarrollarlas.
En la actualidad, no existen datos concluyentes en cuanto al tratamiento preventivo de episodios tromboembólicos en pacientes con insuficiencia cardiaca crónica y dilatación ventricular con diagnóstico de DMD. En casos de miocardiopatía dilatada con fracción de eyección menor del 30%, está recomendado el tratamiento anticoagulante si se asocian fibrilación auricular, trombos intracavitarios o eventos tromboembólicos previos; en ausencia de estos factores asociados, está justificado el tratamiento antiagregante con aspirina2.
Buscamos con este caso destacar la relevancia del reconocimiento, en forma temprana, de un ACV en pacientes con distrofia muscular, lo que llevaría a un manejo más adecuado en la fase aguda y la optimización de la prevención primaria y secundaria de los mismos. Esto contribuiría a disminuir las tasas de morbimortalidad en pacientes que presentan este tipo de miopatías. Entre estas medidas, la utilización de la anticoagulación, teniendo en cuenta el mecanismo cardioembólico en la fisiopatología de estos fenómenos, sea probablemente una terapéutica adecuada, aunque todavía se requiere de más evidencia en el uso de los mismos en este tipo de pacientes para obtener mayor nivel de recomendación.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
