




La distrofia muscular de Emery-Dreifuss (DMED), caracterizada por la tríada clínica de contracturas articulares, debilidad muscular y afectación cardíaca, es genéticamente heterogénea. Causada por mutaciones con herencia ligada al X en los genes EMD y FHL1 y formas dominantes o recesivas, en el gen LMNA.
Caso clínicoPaciente de 41 años con miocardiopatía dilatada y bloqueo auriculoventricular, comienza a los 6 años con lordosis acentuada. Progresivamente agrega debilidad y atrofia en 4 miembros, inicialmente humeroperoneal y luego en cinturas, y retracciones articulares en codos. EMG: compromiso miopático. Biopsia muscular: distrofia muscular, emerina deficiente. Estudio molecular: mutación NM_000117.2:c.461_465dup;p.Tyr155_Gly156InsCTfsX82 en el gen EMD en hemicigosis.
ConclusionesSe describen los hallazgos típicos en la distrofia muscular de Emery-Dreifuss en un caso que presentó una nueva mutación, no reportada previamente. Las características clínicas y antecedentes familiares proporcionan la orientación inicial en el diagnóstico y determinan la solicitud de estudios definitivos.
Emery-Dreifuss muscular dystrophy, characterized by the clinical triad of joint contractures, muscle weakness and cardiac involvement, is genetically heterogeneous. Caused by X-linked mutations in EMD and FHL1 genes, and dominant or recessive forms in the LMNA gene.
Case reportForty-one year old patient with dilated cardiomyopathy and atrioventricular block, begins to have a marked lordosis at the age of 6. Progressively, weakness and atrophy of the 4 limbs, humero-peroneal at first and then at the waist, and joint contractures in elbows become additional symptoms. EMG: myophatic compromise. Muscle biopsy: muscular dystrophy, emerin deficient. Molecular study: NM_000117.2:c.461_465dup;p.Tyr155_Gly156InsCTfsX82 mutation in the EMD gene in hemicigosis.
ConclusionsTypical findings in Emery-Dreifuss muscular dystrophy are described in a case that presented a new mutation, not previously reported. Family history and clinical features provide the initial clues in the diagnosis and determine the request for definitive studies.
La distrofia muscular de Emery-Dreifuss (DMED) se caracteriza por contracturas articulares tempranas en los codos, cuello y tendón de Aquiles, debilidad muscular progresiva humeroperoneal y miocardiopatía asociada con trastornos de la conducción cardíaca1–3
El aspecto más severo de esta enfermedad es el compromiso cardíaco, que usualmente es evidente junto con la progresión de la debilidad muscular, pero puede preceder al compromiso musculo esquelético4–6. La edad de inicio, la severidad y la progresión del compromiso muscular y cardíaco suelen presentar variaciones entre los individuos afectados7.
Los 3 genes cuyas mutaciones son conocidas como causa de la DMED son el EMD (que codifica para emerina) y FHL1 (que codifica para FHL1), ambos relacionados con las formas ligadas al X; y el gen LMNA (que codifica para las proteínas lamina A y C), que determina las formas autosómicas dominantes y recesivas2,3,8,9.
La emerina es una proteína transmembrana, situada en la membrana interna de la envoltura nuclear. Entre sus funciones se conoce un papel en el ensamblaje del núcleo, mantenimiento de la estabilidad de la envoltura nuclear y la regulación de la expresión de ciertos genes. Existe evidencia de su expresión en estadios tempranos de la diferenciación de células musculares3,7,10.
Este reporte describe los hallazgos clínicos, electrofisiológicos, histológicos y moleculares en un paciente con DMED ligada al X producida por una nueva mutación en el EMD, que codifica para emerina.
Caso clínicoPaciente masculino de 41 años con antecedentes de tabaquismo, colecistectomía, miocardiopatía dilatada y bloqueo auriculoventricular que requirió colocación de marcapasos definitivo, derivado por su médica cardióloga al servicio de Neurología para estudio.
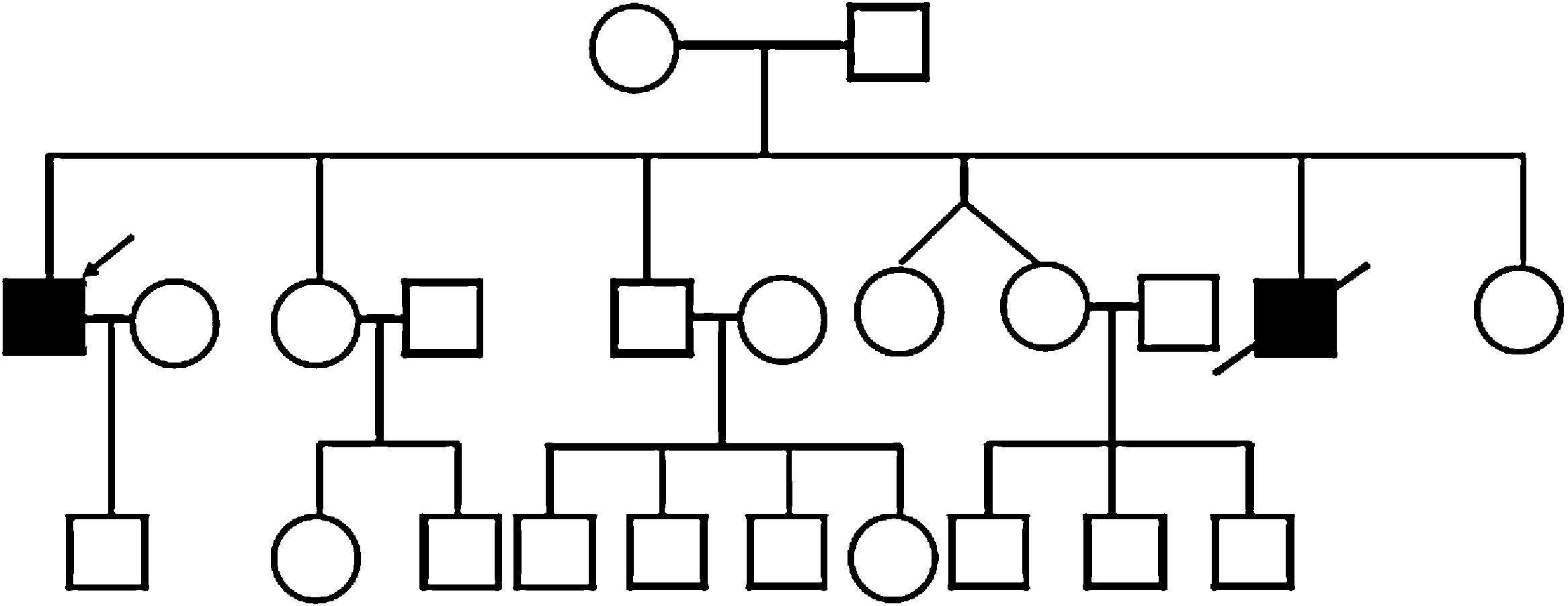
Como antecedentes familiares (fig. 1) presenta un hermano fallecido a los 28 años por complicaciones de una miocardiopatía dilatada no filiada, que al momento de presentarla, según el relato de la madre, tenía contracturas articulares en los miembros superiores.

El paciente comienza a los 6 años aproximadamente con lordosis acentuada. Durante la adolescencia agrega debilidad distal en miembros inferiores y, aproximadamente a los 20 años de edad, la debilidad progresa a nivel proximal de sus miembros superiores.
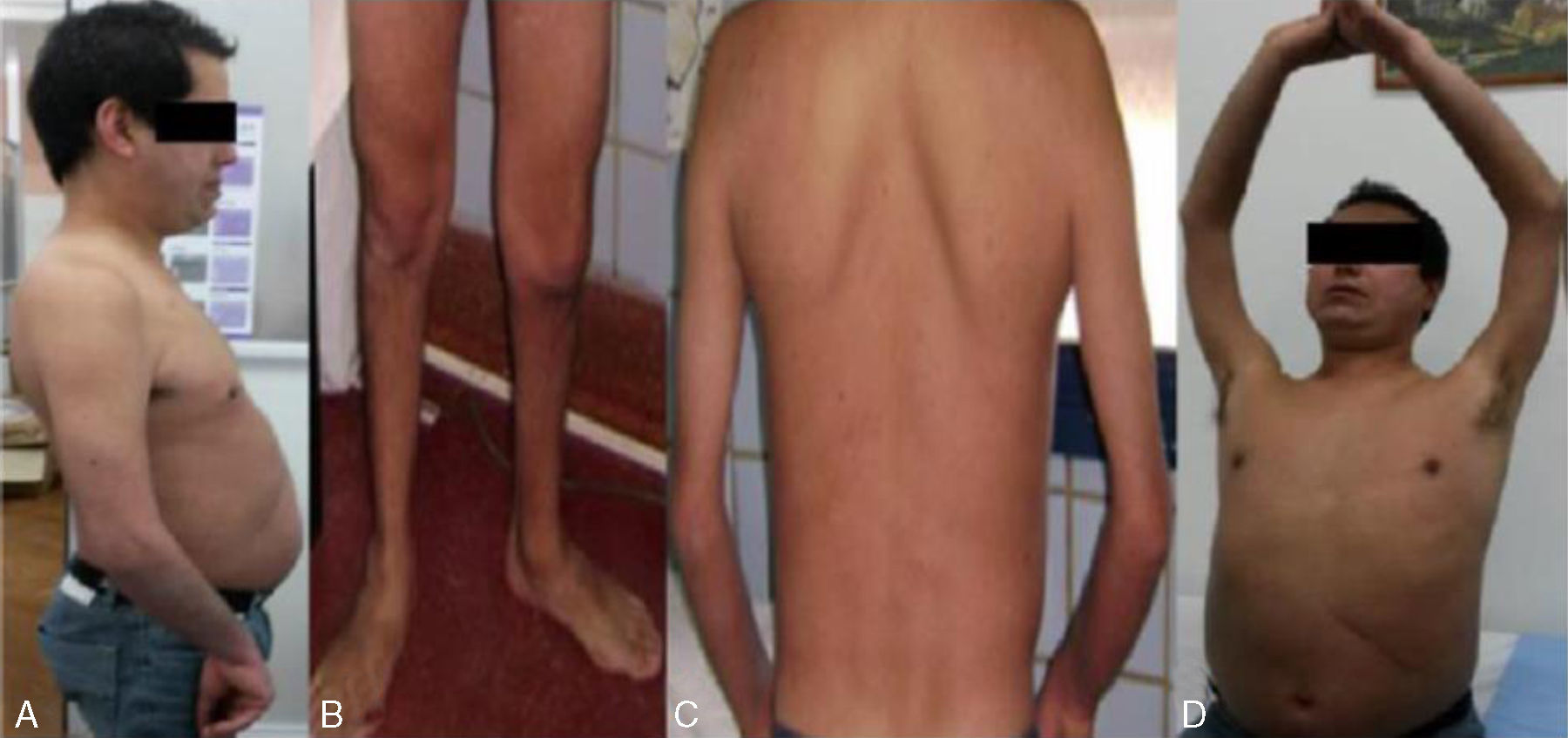
En el examen físico (fig. 2) se evidencia lordosis acentuada y moderada retracción articular en ambos codos. También presenta como hallazgos escápula alada, atrofia muscular a nivel proximal en miembros superiores y peroneal, debilidad proximal en 4 miembros, con predominio de cintura escapular, hiporreflexia generalizada, maniobra de Gowers positiva y una marcha de tipo miopática.

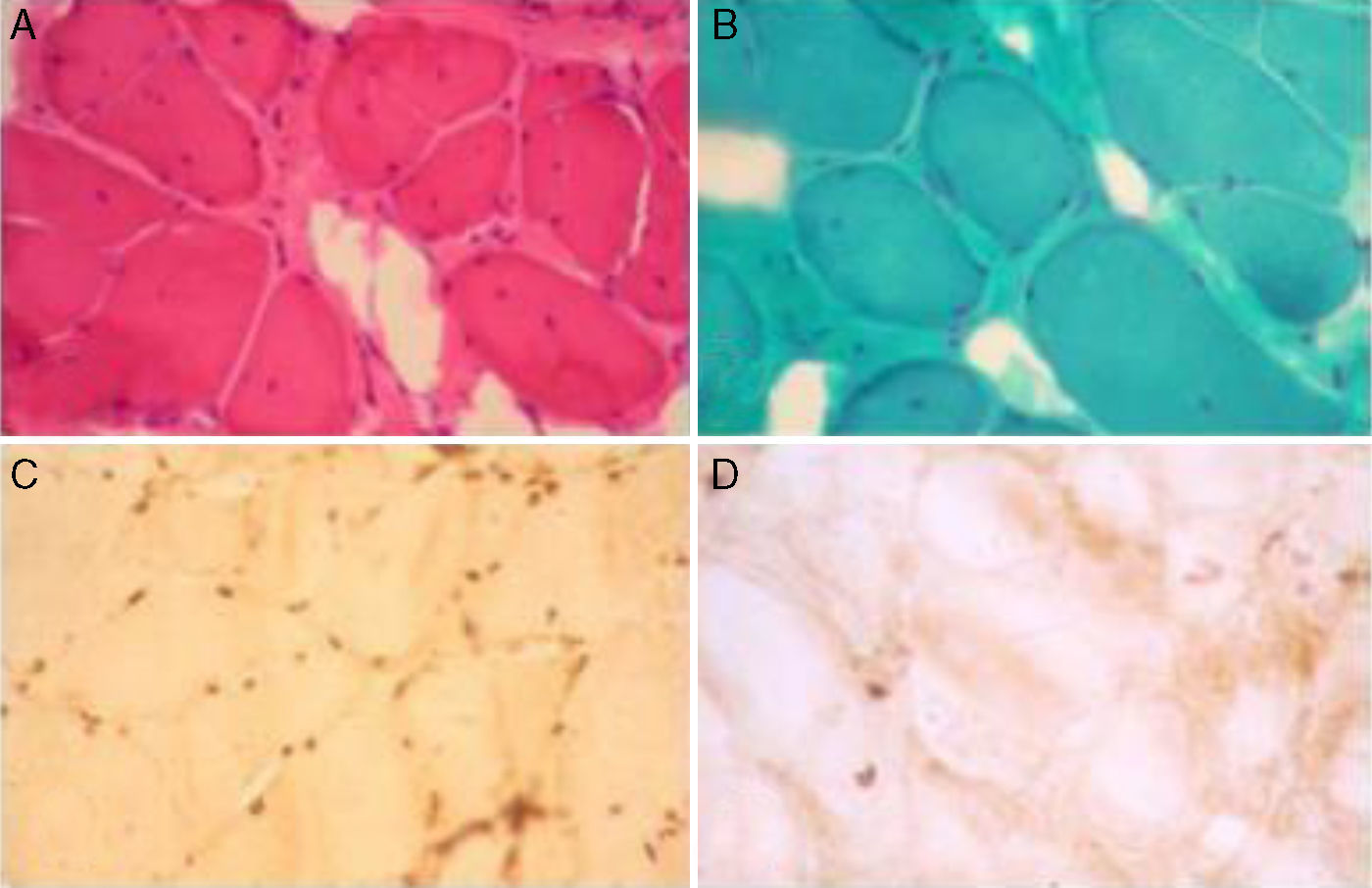
En los estudios complementarios se evidencia un nivel de CPK de 656UI/L (valor de referencia entre 0 y 195UI/L). El ecocardiograma revela dilatación leve del ventrículo izquierdo y moderada de ambas aurículas, con función sistólica y diastólica conservadas, siendo compatible con una miocardiopatía dilatada. Se le realiza un electromiograma que determina compromiso de tipo miopático en 4 miembros. La biopsia muscular de deltoides derecho (fig. 3) evidenció extrema esclerolipomatosis de sustitución, cambios miopáticos crónicos tipo distrófico con predominio histoquímico por probable denervación/reinervación miógena, e inmunomarcación deficiente para emerina.
 hematoxilina-eosina y B) tricrómico de Gomori que evidencian cambios miopáticos crónicos, con lipomatosis interfascicular, fibras redondeadas con atrofia e hipertrofia, partición anormal de los núcleos y algunos núcleos internalizados. C) Control con marcación de emerina en la membrana nuclear. D) Inmunohistoquímica para emerina del paciente, con ausencia de tinción en los núcleos.")
Biopsia muscular de deltoides derecho del paciente: Tinción con A) hematoxilina-eosina y B) tricrómico de Gomori que evidencian cambios miopáticos crónicos, con lipomatosis interfascicular, fibras redondeadas con atrofia e hipertrofia, partición anormal de los núcleos y algunos núcleos internalizados. C) Control con marcación de emerina en la membrana nuclear. D) Inmunohistoquímica para emerina del paciente, con ausencia de tinción en los núcleos.
Tras el informe de la biopsia muscular se confirma la presunción diagnóstica de DMED.
En los controles clínicos posteriores se evidencia empeoramiento en su debilidad de cinturas, agregando debilidad distal en miembros superiores y reflejos osteotendinosos ausentes en forma generalizada. En este contexto, el paciente refiere mayor dificultad para levantar los brazos y ponerse de pie.
Posteriormente es derivado para realizarse un estudio molecular. Mediante el análisis de región codificante de gen EMD mediante PCR-secuenciación por Sanger por electroforesis capilar se evidenció la presencia de la mutación NM_000117.2:c.461_465dup;p.Tyr155_Gly156InsCTfsX82 en el gen EMD en hemicigosis (cromosoma X). La misma causa un corrimiento en el marco de lectura y la aparición de un codón de stop prematuro 82 codones río abajo. Si bien no ha sido previamente descripta como causa de Emery-Dreifuss, se puede predecir con alto grado de confianza su potencialidad deletérea sobre la emerina y su consecuente patogenicidad en el trastorno que afecta al paciente estudiado.
Fueron estudiadas la madre y una hermana, que resultaron no ser portadoras de la mutación en ADN purificado de leucocitos, por lo que, dada la probable coexistencia de la enfermedad en el hermano del paciente presentado, se puede asumir que la madre probablemente presente mosaicismo gonadal o de la línea germinal para la mutación causante.
ComentariosEl diagnóstico de todas las formas de DMED se basa en los hallazgos clínicos y la historia familiar. En los casos de herencia ligada al X por mutación del gen de emerina se requiere también la ausencia de emerina en la biopsia muscular y posteriormente el estudio molecular2,11–13. En los casos de la mutación en el gen FHL1, dado que esta forma de FHL1 no presenta cuerpos reductores14, generalmente se confirma directamente mediante el estudio molecular cuando, frente a un fenotipo de Emery-Dreifuss, la emerina está preservada y no existe deficiencia de lamina. Eventualmente se puede hacer también por inmunofluorescencia o Western Blot. En las formas autosómicas dominantes y recesivas, se basa en el estudio molecular del gen LMNA2,7,15.
Este caso es compatible con el cuadro clínico típico de un paciente con Emery-Dreifuss: la atrofia muscular humeroperoneal, las contracturas articulares a nivel de los codos así como la debilidad en la cintura escapular y pelviana, junto a la miocardiopatía dilatada y el trastorno de la conducción cardíaca16. Los hallazgos descriptos en su hermano y la ausencia de emerina en la inmunohistoquímica de la biopsia muscular orientaron a dirigir el estudio molecular hacia el gen de EMD, de cuyo estudio surgió la presencia de la mutación NM_000117.2:c.461_465dup;p.Tyr155_Gly156InsCTfsX82 en hemicigosis (cromosoma X), no reportada hasta la fecha. Se requirió de un manejo interdisciplinario para arribar al diagnóstico. La terapéutica actual se basa en limitar las deformidades, conservar la función cardíaca y realizar un adecuado control del marcapasos, disminuyendo el riesgo de muerte súbita y aumentando la supervivencia. La adecuada caracterización de estos pacientes, además de brindar información y las herramientas para un adecuado asesoramiento genético, constituyen la clave para profundizar las investigaciones en terapias modificadoras de la enfermedad.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
