





La enfermedad de Charcot-Marie-Tooth (CMT) es la polineuropatía sensitivo-motora hereditaria más frecuente en la práctica médica. Su sospecha diagnóstica se hace en pacientes con hallazgos clínicos del sistema nervioso periférico, con atrofia de extremidades superiores e inferiores, trastornos sensitivos en guante y calcetín, arreflexia osteotendinosa generalizada y hallazgos electrofisiológicos compatibles. Es infrecuente que la clínica sea afectación de nervios craneanos, aunque se han reportado casos de concomitancia de miastenia gravis ocular. Existen algunos reportes asociados a parkinsonismo y disfunción de nervios craneanos, cuadros de oftalmoplejía familiar y CMT, y otros aislados de neuropatía craneal monofásica o múltiple. Reportamos 2 pacientes con neuropatía craneal múltiple, quienes tenían hallazgos clínicos y electrofisiológicos de CMT, haciendo énfasis en que es una presentación clínica poco habitual, pero a tener en cuenta.
Charcot-Marie-Tooth disease (CMT) is the most common hereditary sensorimotor polyneuropathy in clinical practice. The diagnosis must be suspected in patients with clinical findings at the peripheral nervous system, like upper- and lower-limb atrophy, sensory “stocking-glove” disorders, generalized loss of stretch or myotatic reflex and compatible electrophysiological findings. Cranial nerves involvement is rare, although there have been reports of concomitance with ocular myasthenia gravis, and some reports associated with parkinsonism and cranial nerve dysfunction, or family ophthalmoplegia and CMT, and isolated monophasic or multiple cranial neuropathy. We report the cases of 2 patients with multiple cranial neuropathy who had clinical and electrophysiological findings of CMT-1A, emphasizing that –although it is a rare clinical presentation–, the diagnosis must be considered in certain cases.
La enfermedad de Charcot-Marie-Tooth (CMT), también llamada neuropatía sensitivo-motora hereditaria, fue descrita en forma independiente por Charcot y Marie en París y por Tooth en Londres en 1866. Se transmite de forma autosómica dominante en la mayoría de los casos, aunque también de forma autosómica recesiva o ligada a X. Tiene una incidencia estimada de 1/2.500 nacidos vivos, y una prevalencia de 17 a 25 por 100.000 habitantes, siendo el desorden neuromuscular hereditario más común. Su penetrancia es incompleta, y tiene heterogeneidad genética y expresividad variable1,2. En relación con lo anterior, el espectro de genes descubiertos hasta la fecha que pueden causar CMT continúa ampliándose, con más de 1.000 mutaciones diferentes en 80 o más genes, expresándose en presentaciones clínicas variadas3. Sin embargo, el fenotipo clásico es el de una neuropatía motora distal simétrica, lentamente progresiva, que afecta las extremidades superiores y las extremidades inferiores (EEII) generando debilidad y atrofia de musculatura distal de las manos y los pies, hipotrofia tenar, mano en garra y aumento del arco plantar, dedos en martillo y marcha equina, también llamada en steppage. A esto se suma arreflexia osteotendínea (principalmente aquiliana), hipoestesia distal con compromiso de sensibilidad vibratoria, táctil y dolorosa en las 4 extremidades y disminución de sensibilidad propioceptiva, lo cual puede generar ataxia. Otros síntomas asociados pueden incluir deformidad ósea, escoliosis, nervios palpables (hipertrofiados) en el 20-25% de los casos (principalmente el nervio auricular mayor, el nervio cubital y el nervio peroneo lateral)4 y ocasionalmente afectación de nervios craneales5,6. La edad de inicio de los síntomas, en la gran mayoría de los casos, se presentan entre la primera y la segunda década, sin embargo existen subtipos de CMT con presentación clínica en la cuarta-quinta década de la vida, constituyendo estos últimos casos esporádicos y excepcionales7.
La clasificación de la enfermedad de CMT se basa en patrones de herencia y genética molecular, apoyados en resultados de estudio electrofisiológicos y de biopsia de nervio. La neurología clásica la divide en 2 grandes grupos; la tipo 1 que corresponde a las variante desmielinizante, la tipo 2 o variante axonal, así como signos de degeneración y regeneración axonal crónica en biopsia de nervio. Sumado a lo anterior, la clasificación según la genética molecular determina 6 grupos específicos que no corresponde especificar al presente artículo, pero que pueden consultarse en las referencias1,2,7.
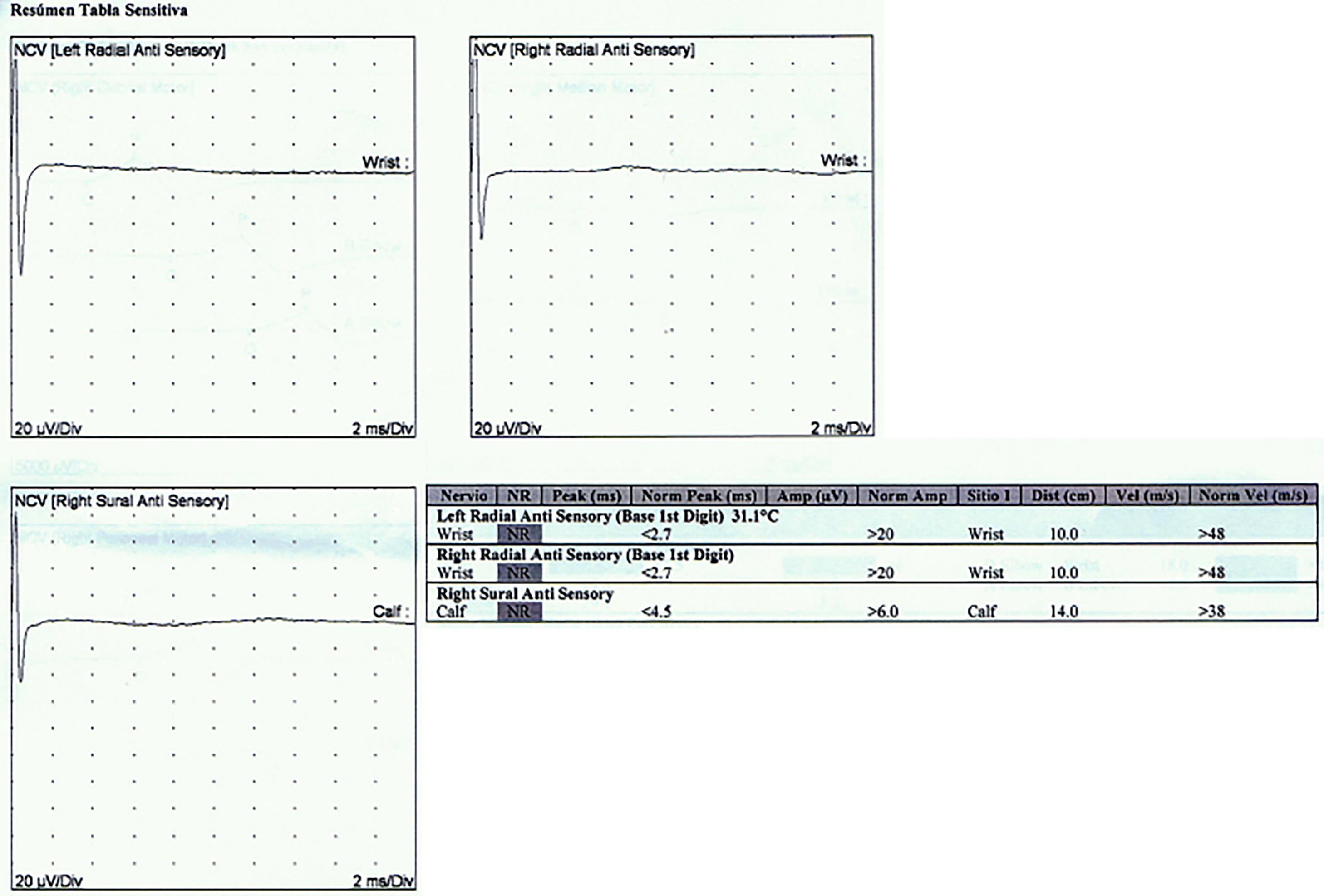
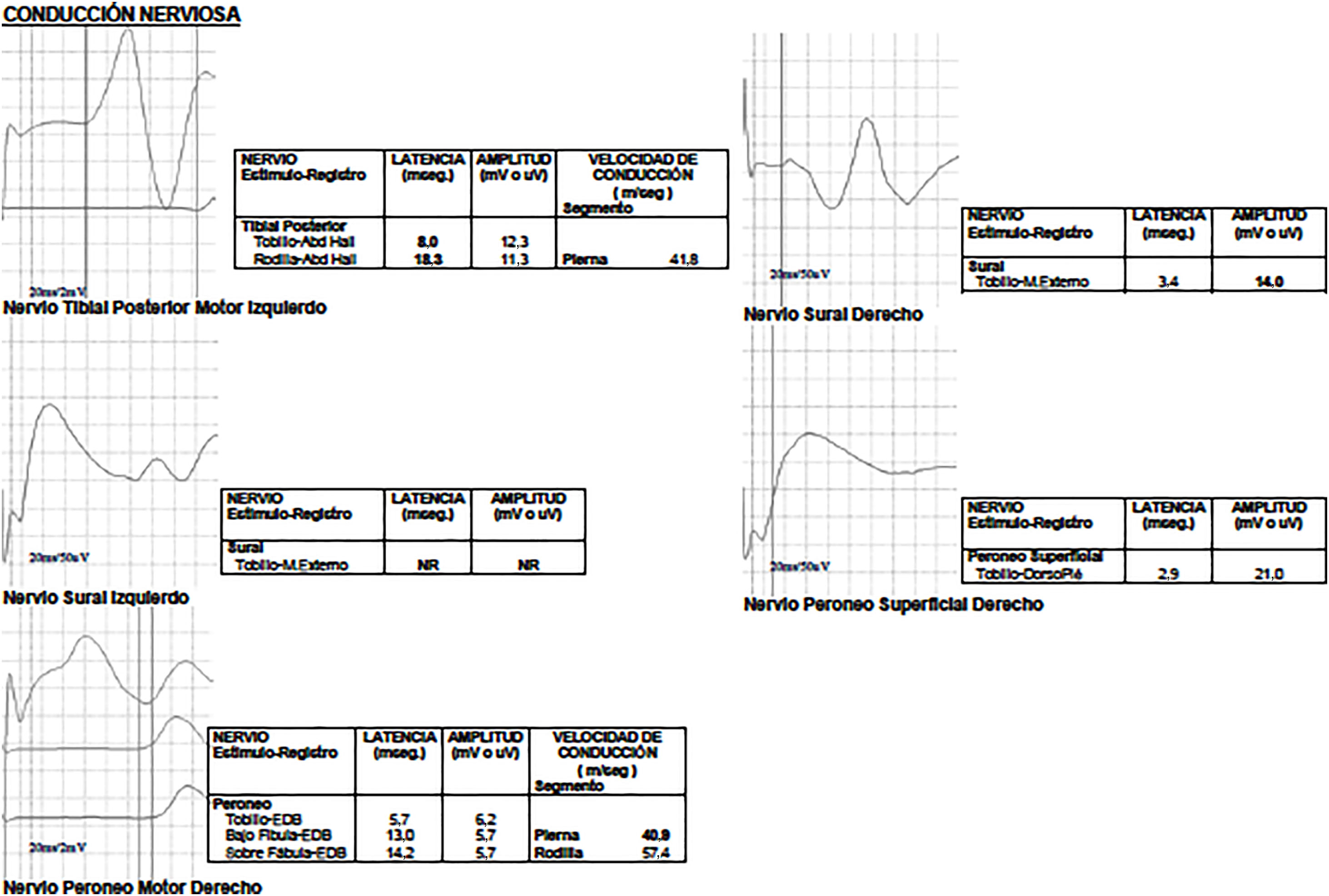
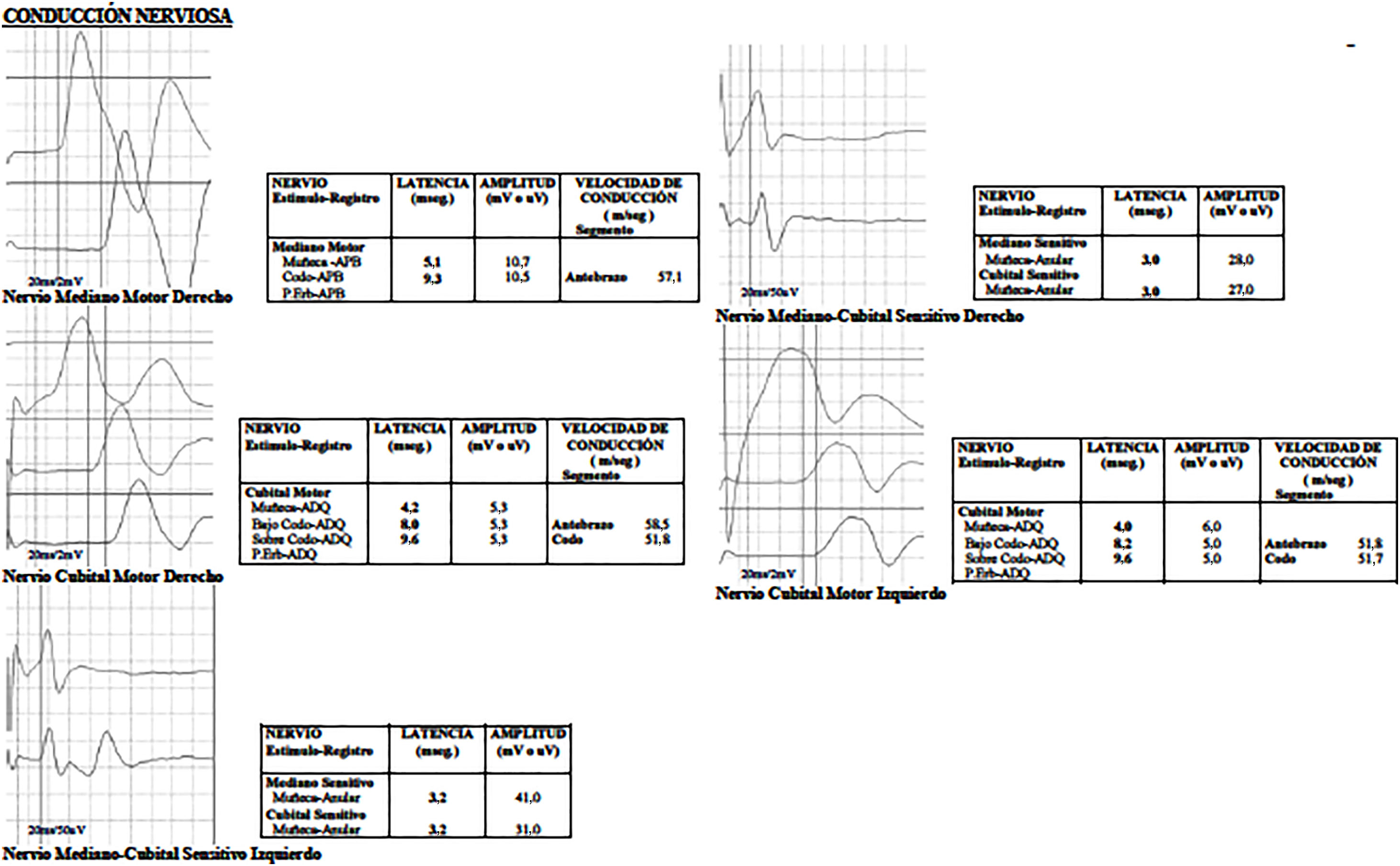
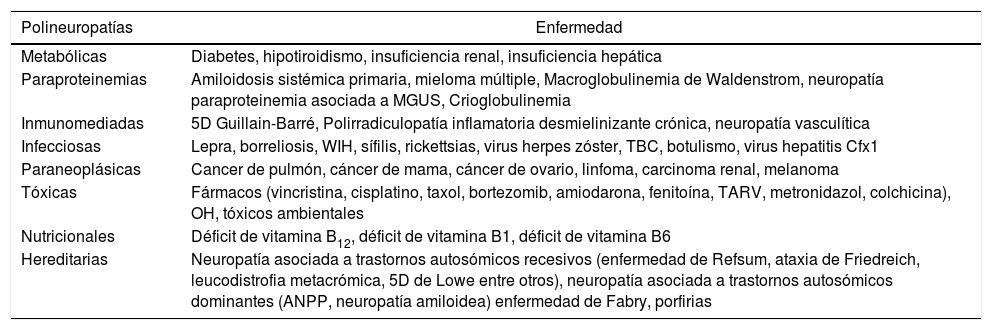
Idealmente, el diagnóstico se basa en una historia familiar —aunque esta puede ser negativa debido a múltiples razones—, como la expresión subclínica, la herencia autosómica recesiva o la mutación de novo de un gen dominante1,2; en estos casos, hay que excluir otro origen de polineuropatías como las enunciadas en la tabla 18–10. A lo anterior hay que sumar estudios electrofisiológicos (EMG y VCN) que cuando se realizan de buena forma y se relacionan con la clínica sugerente pueden constituir el sustento del diagnóstico, que se basa en un patrón desmielinizante simétrico de las extremidades superiores e inferiores, con latencias distales prolongadas, velocidades de conducción <38m/seg, y potenciales de acción muscular compuesto relativamente conservados en amplitud, sin franca dispersión temporal (CMT-1A); y por otro lado, en un patrón axonal simétrico con latencias y velocidades de conducción conservadas, asociadas a caída de las amplitudes de los potenciales de acción muscular compuesto (CMT-2)11. Así mismo, también se apoya en la biopsia de nervio (habitualmente el sural), que típicamente muestra cambios de una neuropatía hipertrófica, con la formación de «bulbo de cebolla», lo que sin embargo no es un signo específico de este tipo de polineuropatías y actualmente está reservada para pacientes en los que existe una sospecha clínica de una neuropatía inflamatoria adquirida y potencialmente tratable7. Y, finalmente, los estudios genéticos recomendados según lo establecido en la literatura7.
Principales etiologías de las polineuropatías
| Polineuropatías | Enfermedad |
|---|---|
| Metabólicas | Diabetes, hipotiroidismo, insuficiencia renal, insuficiencia hepática |
| Paraproteinemias | Amiloidosis sistémica primaria, mieloma múltiple, Macroglobulinemia de Waldenstrom, neuropatía paraproteinemia asociada a MGUS, Crioglobulinemia |
| Inmunomediadas | 5D Guillain-Barré, Polirradiculopatía inflamatoria desmielinizante crónica, neuropatía vasculítica |
| Infecciosas | Lepra, borreliosis, WIH, sífilis, rickettsias, virus herpes zóster, TBC, botulismo, virus hepatitis Cfx1 |
| Paraneoplásicas | Cancer de pulmón, cáncer de mama, cáncer de ovario, linfoma, carcinoma renal, melanoma |
| Tóxicas | Fármacos (vincristina, cisplatino, taxol, bortezomib, amiodarona, fenitoína, TARV, metronidazol, colchicina), OH, tóxicos ambientales |
| Nutricionales | Déficit de vitamina B12, déficit de vitamina B1, déficit de vitamina B6 |
| Hereditarias | Neuropatía asociada a trastornos autosómicos recesivos (enfermedad de Refsum, ataxia de Friedreich, leucodistrofia metacrómica, 5D de Lowe entre otros), neuropatía asociada a trastornos autosómicos dominantes (ANPP, neuropatía amiloidea) enfermedad de Fabry, porfirias |
Tiene también lugar los estudios imagenológicos, como la resonancia magnética nuclear (RMN), que es de utilidad al permitir evaluar los segmentos proximales del plexo lumbosacro y los nervios craneales que muestran habitualmente engrosamiento de uno o más nervios o plexos nerviosos asociado a cambios denervativos en la musculatura distal, o a través de ecotomografía neuromuscular, evidenciando engrosamiento de los nervios periféricos de tipo uniforme y difusa, contrastando con el engrosamiento desigual y focal observado en las neuropatías desmielinizantes adquiridas7.
Actualmente no hay un tratamiento curativo, por lo que se sugiere terapia física oportuna con el fin de prevenir o disminuir la aparición de deformidades, el uso de férulas para fortalecer la musculatura y mejorar la deambulación y ejercicios de bajo impacto, tales como la natación. En caso de deformidades óseas la cirugía es una alternativa6. En la actualidad existen una serie de ensayos en los cuales se ha utilizado ácido ascórbico para CMT1A, basado en un modelo animal de CMT1A (ratones transgénicos que sobreexpresaron PMP22) observándose una inhibición en la expresión de PMP22 en dichos casos; sin embargo, en seres humanos aún no se han mostrado beneficios consistentes de dicha terapia. Por otra parte, existen investigaciones en fase i y ii que evalúan la administración de coenzima Q-10 a dosis de 300mg 2 veces al día por 3 meses, observándose una reducción significativa del dolor, debilidad muscular y fatiga2,3. En este contexto el uso de paracetamol y AINE pueden ser útiles en el alivio del dolor musculoesquelético, así como también los antidepresivos tricíclicos, la carbamazepina y la gabapentina si hay dolor neuropático asociado. Algunos reportes describen también una discreta mejoría en pacientes con CMT1B después del tratamiento con corticoides y antioxidantes, así como en pacientes con CMT tratados con inmunoglobulinas y plasmaféresis. Por otro lado, es importante para los pacientes con CMT evitar el uso de neurotoxinas, como alcohol y drogas2.
Debido a que el compromiso de los pares craneanos en CMT es poco frecuente, presentamos a continuación 2 casos clínicos en los cuales esta fue la clínica predominante.
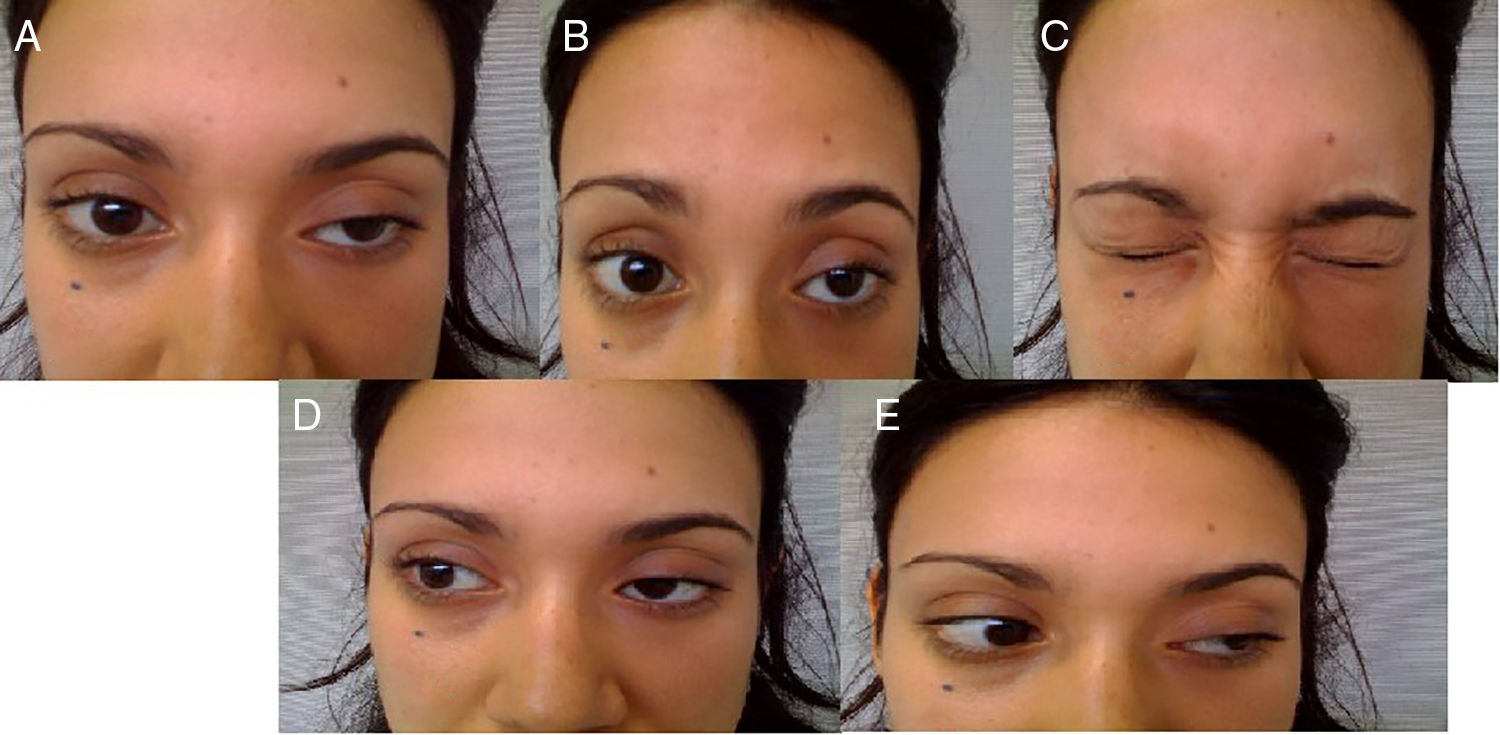
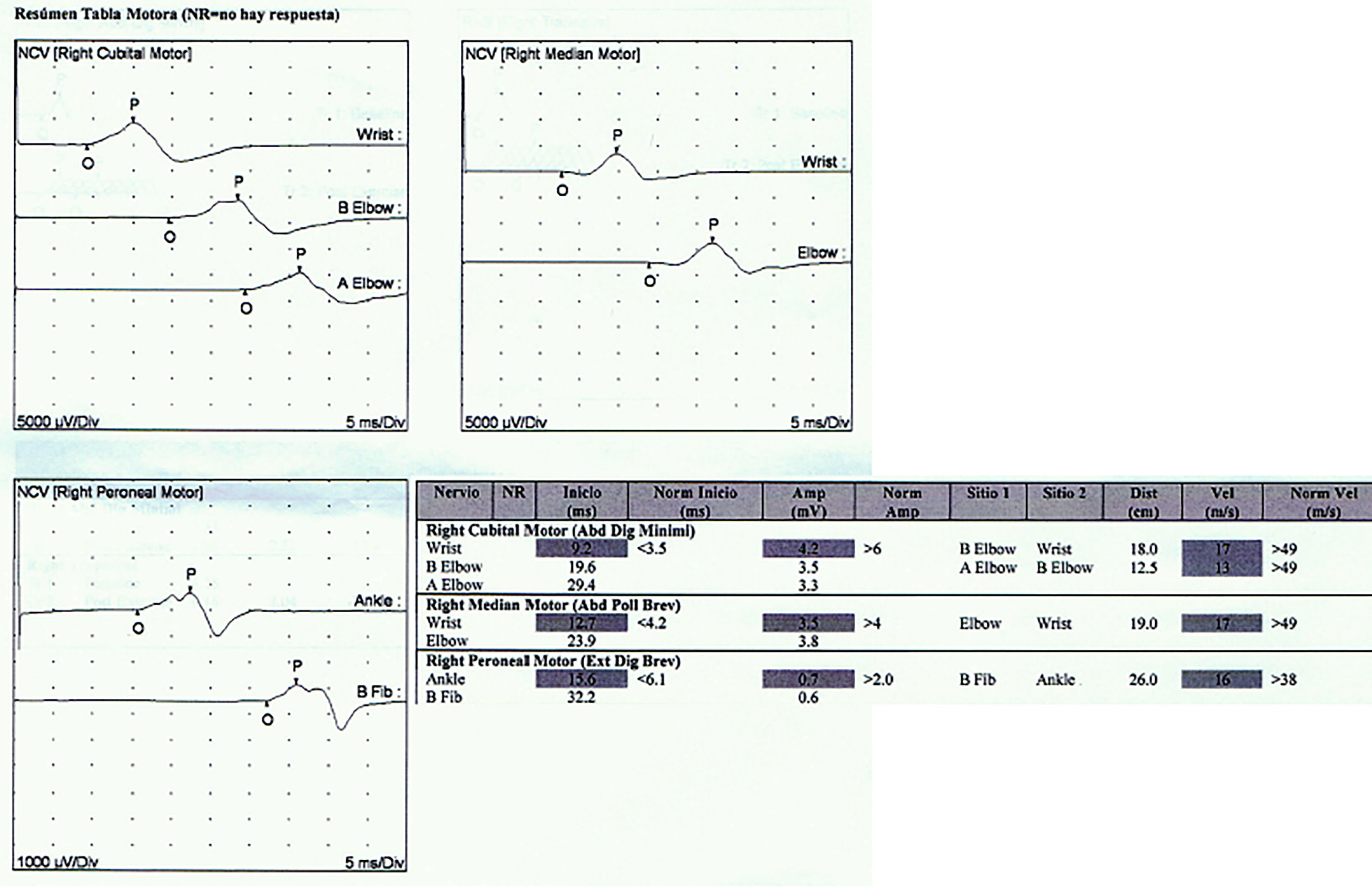
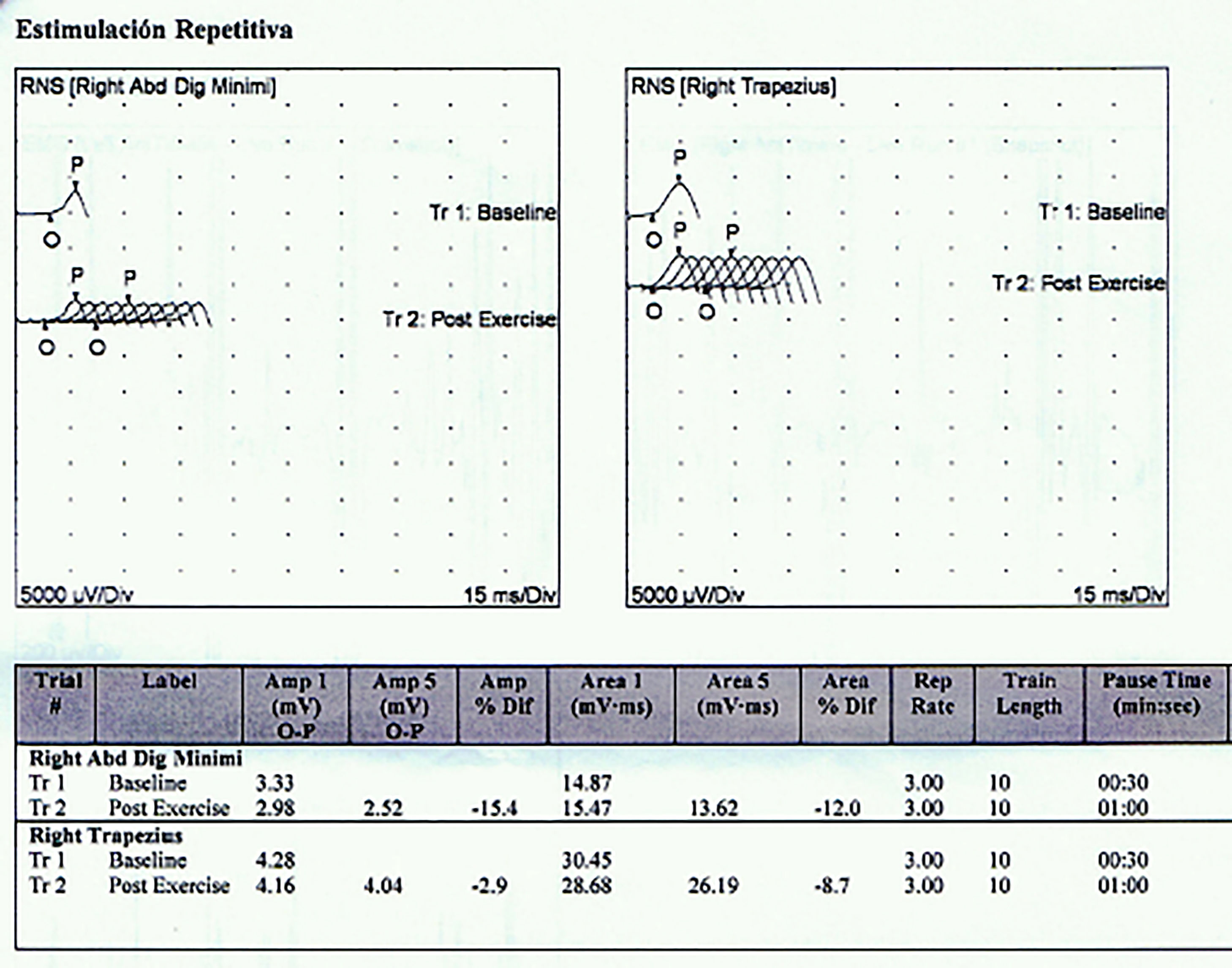
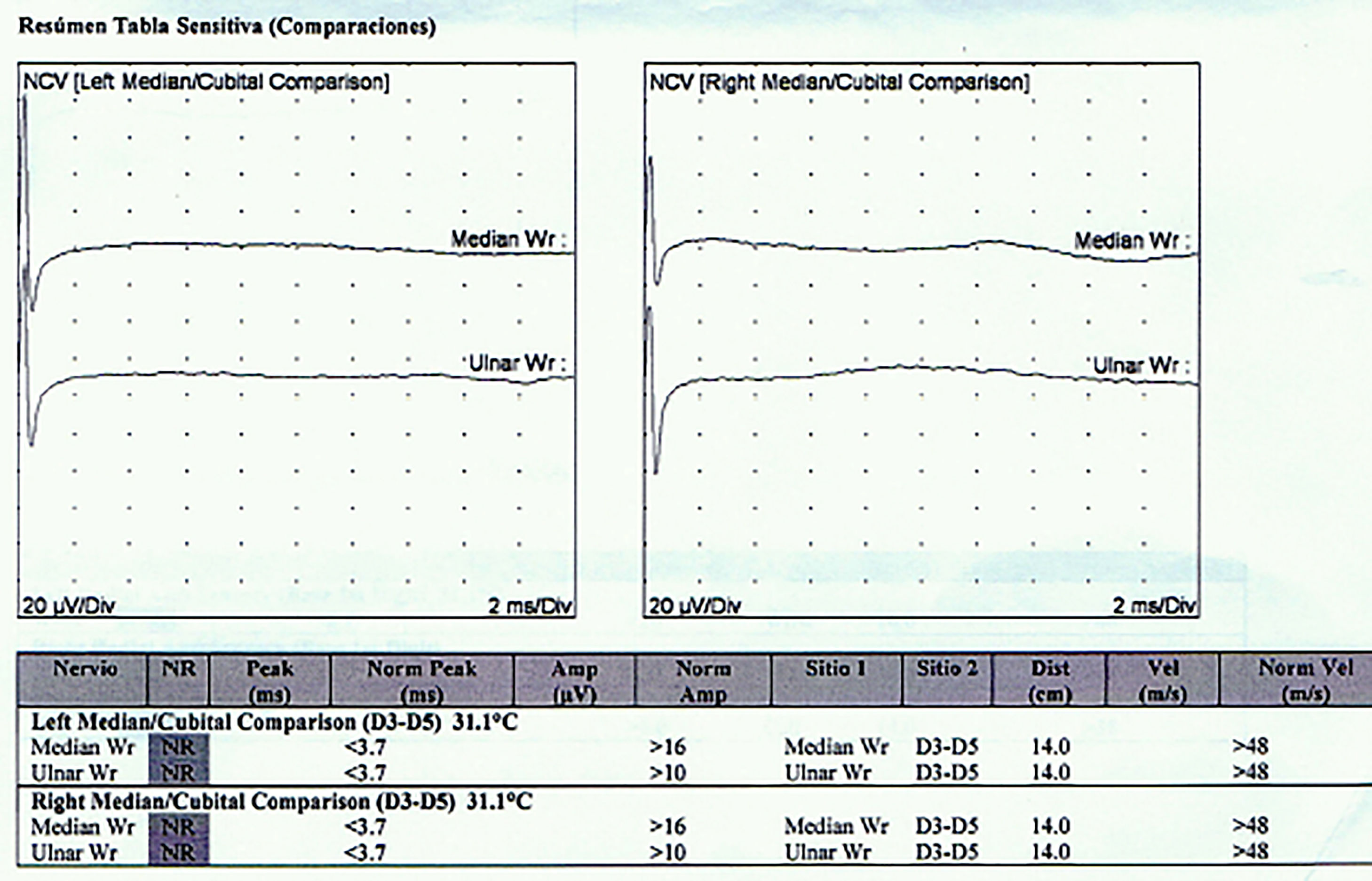
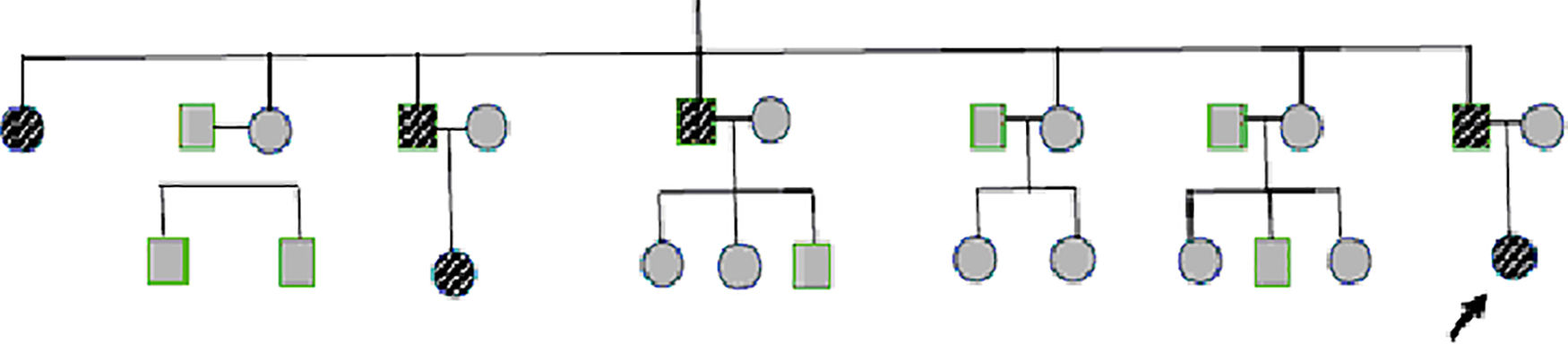
Reporte de casosCaso 1Mujer de 21 años de edad, sin antecedentes mórbidos, con historia de un año de evolución de instalación lenta y progresiva de ptosis palpebral de predominio a izquierda, asociado a visión doble, síntomas de curso fluctuante, de mayor intensidad vespertina, sin disfagia ni disartria. Fue evaluada por oftalmología y derivada a consulta neurológica. Al examen físico neurológico destacaba ptosis bipalpebral, de mayor severidad a izquierda, asociada a paresia del recto lateral y medial derechos y reducción de los pliegues frontales, y paresia leve de ambos orbiculares de los párpados (fig. 1). No se obtuvo aumento de los hallazgos a la prueba de fatigabilidad ptósica ni a la maniobra de Marie-Walker. Se indicó una RMN cerebral, reportada como normal, y se practicó un estudio de conducción nerviosa, con test de estimulación repetitiva y electromiografía de 4 extremidades, donde se descarta un defecto de la unión neuromuscular y se confirman hallazgos electrofisiológicos de una neuropatía hereditaria tipo Charcot-Marie-Tooth tipo I-A (figs. 2-5). Se complementa con tomografía de tórax y mediastino, que es normal. Al interrogatorio dirigido la paciente relata historia familiar positiva (fig. 6).






Mujer de 17 años de edad, sin antecedentes mórbidos relevantes, quien consulta por historia que parte en la niñez de caídas frecuentes, torpeza motora y parestesias fluctuantes de las extremidades superiores e inferiores. Durante su evolución se presenta 3 meses previos a la consulta neurológica dolor tipo neurálgico en distribución ulnar derecha, por lo que traumatología, ante la sospecha de atrapamiento ulnar en el codo, solicita estudio electrofisiológico que muestra hallazgos electrofisiológicos de una neuropatía hereditaria tipo Charcot-Marie-Tooth tipo I-A (figs. 7 y 8). A la evaluación en la unidad de neurología se evidencia una ptosis palpebral izquierda, no fluctuante ni que empeora a la prueba de fatigabilidad ptósica ni a la maniobra de Marie-Walker, y diparesia facial discreta con reducción de los pliegues frontales y paresia del cierre bipalpebrala.


La enfermedad de CMT corresponde a un grupo de enfermedades neuromusculares hereditarias con presentaciones clínicas y anormalidades heterogéneas12. La enfermedad de CMT asociada a compromiso de pares craneanos es una manifestación clínica inhabitual que se puede observar, y que infrecuentemente se describe en la literatura5. De esta forma se presentan los casos mencionados, existiendo compromiso del iii, vi y vii par en el caso 1 y compromiso del iii y vii par en el caso 2, constituyendo en sí un claro ejemplo de neuropatías craneales múltiples. Las neuropatías craneales múltiples son comunes en neurología, con etiologías que van desde las relativamente benignas y tratables hasta las etiologías francamente malignas y que amenazan la vida del paciente (tabla 2), y constituyen un desafío diagnóstico formidable, ya que los nervios craneales pueden afectarse en cualquier sitio a lo largo de su trayecto, desde el tronco encefálico hasta su trayecto extramedular. El diagnóstico diferencial es extenso, siendo clave considerar la edad del paciente, su estado inmunocompetente y la progresión del compromiso neurológico para enfocar de esta forma el estudio inicial más apropiado13.
Diagnóstico diferencial etiológico de neuropatías craneales múltiples
| Neoplásico | Schawnnoma, meningioma, metástasis, linfoma, carcinoma nasofaríngeo, adenoma pituitario, leucemia, mieloma, entre otros |
| Vascular | ACY, complicación quirúrgica, enfermedad desmielinizante (esclerosis múltiple, Guillain-Barré, 5D Miller-Fisher, polineuropatía inflamatoria desmielinizante crónica), toxicidad de quimioterapia |
| Infección | Lyme, Mycobacterium tuberculosis, pseudomonas, sífilis, botulismo, difteria, listeria, criptococosis, apergilosis, mucormicosis, herpes zoster, CMV, WEB, VIH, Chagas, entre otros |
| Enfermedad sistémica | Sarcoidosis, Becet, amiloidosis, síndrome de Tolosa-Hunt, síndrome Melkersson- Rosenthal, histiocitosis, granulomatosis de Wegener, arteritis de la temporal, poliarteritis nodosa, Sjogren, esclerodermia, LES, Paget, entre otras |
| Otros desórdenes neurológicos | Hipertensión intracraneana idiopática, encefalopatía de Wernicke, polineuropatía craneal idiopática, miastenia gravis, distrofia muscular oculofaríngea, distrofia facioescapulohumeral, síndromes congénitos (p. ej.: Moebius, Charcot-Marie-Toot), entre otros |
La importancia de nuestros casos descritos es que constituyen una neuropatía craneal múltiple como manifestación principal (caso 1), o concomitante (caso 2) asociado a una condición neurológica hereditaria, en este caso CMT, lo cual es poco frecuente, existiendo solo algunos reportes de casos descritos en la literatura. Das et al.14 publicaron el caso de una mujer de 27 años con compromiso bilateral del v y vii par craneano, demostrado por ensachamiento de estos en la RMN, asociado a mutación missense del gen PMP22. Otro caso de un niño de 14 años con compromiso de ix y xii par craneano asociado a duplicación del gen PMP 22 fue reportado por Kulkarni et al.15. Aho et al.16 reportó el caso de un hombre de 64 años con sordera bilateral y dolor facial derecho con estudio genético que demostró una deleción de novo del gen PMP22. Además, se encontró dentro de la literatura una publicación de 1963 por Schwartz et al.17, en donde se describía miembros de una familia con clínica de CMT asociado a oftalmoplejía. De la misma manera, se han descrito casos de CMT asociado a miastenia gravis, con fluctuación ocular de mayor severidad18, o casos tan raros como compromiso de los nervios hipogloso y glosofaríngeo, como fue reportado en un paciente varón de 14 años de edad en Mombay-India15; o la familia con CMT, ptosis, demencia y parkinsonismo que podría explicarse por una degeneración multisitémica que compromete los núcleos y tractos centrales, e hipoventilación crónica que subyace tanto los procesos centrales como periféricos13. La mayoría de estas descripciones inhabituales de compromiso de pares craneanos son descritas en paciente con mutación de PMP22, que es la mutación más frecuente asociada a CMT19; sin embargo, existen subtipos de CMT muy inhabituales, como por ejemplo el 4B1 (1% de todos los CMT4), que se asocia a mutaciones del MTMR2 y en donde no es infrecuente observar, por ejemplo, debilidad facial20.
En los casos reportados el caso 1 clínicamente no mostraba compromiso nervioso periférico evidente, sí tenía el antecedente de familiares con neuropatías hereditarias y se confirmó la presencia de compromiso periférico por hallazgos sugerentes de neuropatía hereditaria tipo CMT tipo 1 en la electromiografía. En el segundo caso la paciente también presentó, asociado al compromiso de pares craneanos, electromiografía de las 4 extremidades con patrón de polineuropatía desmielinizante de una CMT tipo 1. Sin embargo, en ambos casos no se realizó estudio genético, lo que pudiese haber sido de utilidad en cuanto a consejería genética tanto para las pacientes como para sus familiares, y de esta forma identificar la presencia de alguna mutación que se asociase a compromiso de pares craneanos.
En referencia al diagnóstico diferencial enfocado como neuropatía craneal múltiple, en ambos casos no existía antecedente de síntomas B que orientasen a etiología neoplásica, tampoco hallazgos cerebrovasculares por anamnesis y evolución (con imagen negativa en el primer caso). En cuanto a vasculitis y conectivopatías no existía historia de poliartralgias, fiebre, compromiso cutáneo ni pulmonar asociado, tampoco clínica compatible con difteria o viajes que pudiesen haber hecho sospechar enfermedad de Lyme. Igualmente, no había sustento por tiempo de evolución y presentación de cuadro sugerente de Guillain-Barré o sus variantes. Así mismo, y orientándose a etiologías infecciosas, no existía cuadro de adenopatías, exantema ni signos sugerentes que hicieran sospechar infección asociada. Por último, desde el punto de vista clínico y electrofisiológico, se descarta el origen de enfermedad de unión neuromuscular, tanto por la negatividad de la maniobra de Mary Walker como por el test de estimulación repetitiva (respectivamente), y tampoco impresionaba que pudiese haber sido secundario a botulismo, dado que la presentación del cuadro fue mucho más larvado en ambos casos y tampoco existía el antecedente de posibles alimentos contaminados.
ConclusiónDentro de las etiologías de neuropatía craneal múltiple, si bien la enfermedad de CMT es una causa poco frecuente, es importante conocerla, siendo el compromiso nervioso periférico de predominio motor asociado a esta clínica una de las claves para poder sospecharlo, que hay que evidenciar tanto por clínica como por estudios electrofisiológicos, por lo tanto hay que considerarla dentro de las posibles hipótesis diagnósticas una vez descartadas otras etiologías más frecuentes (infecciosas, vasculares y neoplásicas).
Cabe recalcar el importante rol que cumple la electromiografía en el diagnóstico de dicha enfermedad, siendo la herramienta que nos permitió realizar el diagnóstico en ambos casos.
Conflicto de interesesNo hay conflicto de intereses.
Los autores agradecen especialmente a los pacientes, quienesnos brindaron su confianza y generosidad, y a las autoridadesuniversitarias e institucionales que apoyaron esta publicación.
