




La gliomatosis leptomeníngea en ausencia de tumor primario intracerebral es infrecuente. Se asocia a tejido heterotópico glial en el espacio subaracnoideo.
Caso clínicoMujer de 23 años con episodios confusionales. La resonancia magnética mostró lesión en cisterna supraselar derecha, sin realce tras la administración de gadolinio. Se confirmó en forma intraoperatoria la localización extraaxial de la masa. El análisis histopatológico reveló oligoastrocitoma de bajo grado.
ConclusiónA pesar de ser inusual, debe considerarse como diagnóstico diferencial la gliomatosis primaria en masas a nivel del espacio subaracnoideo.
Leptomeningeal gliomatosis without a primary parenchymal involvement is a rare condition associated with heterotopic nests of glial tissue in the subarachnoid space.
Case ReportA 23 years old woman presented with confusional episodes. Magnetic Resonance Imaging showed mass on the right suprasellar cistern, without enhancement after the administration of gadolinium. Surgery stated the extraaxial localization of the mass. The histopathologic analysis revealed low grade Oligoastrocitoma.
ConclusionDespite being unusual, primary leptomeningeal gliomatosis should be considered as a differential diagnosis in masses on subarachnoid space.
Paciente de sexo femenino de 23 años de edad, sin antecedentes de relevancia, que consultó por episodios confusionales leves, asociados a vómitos y amnesia retrógrada reciente. Refirió un cuadro similar unos meses previos a la consulta, con tomografía computarizada (TC) de cerebro normal. El examen neurológico no arrojó alteraciones.
Se le solicitó electroencefalograma (EEG) que reveló descargas breves generalizadas aisladas y otras focales infrecuentes parietotemporales, interpretadas como probables crisis parciales complejas breves o parciales simples con síntomas psíquicos, por lo que se medicó con antiepilépticos (topiramato).
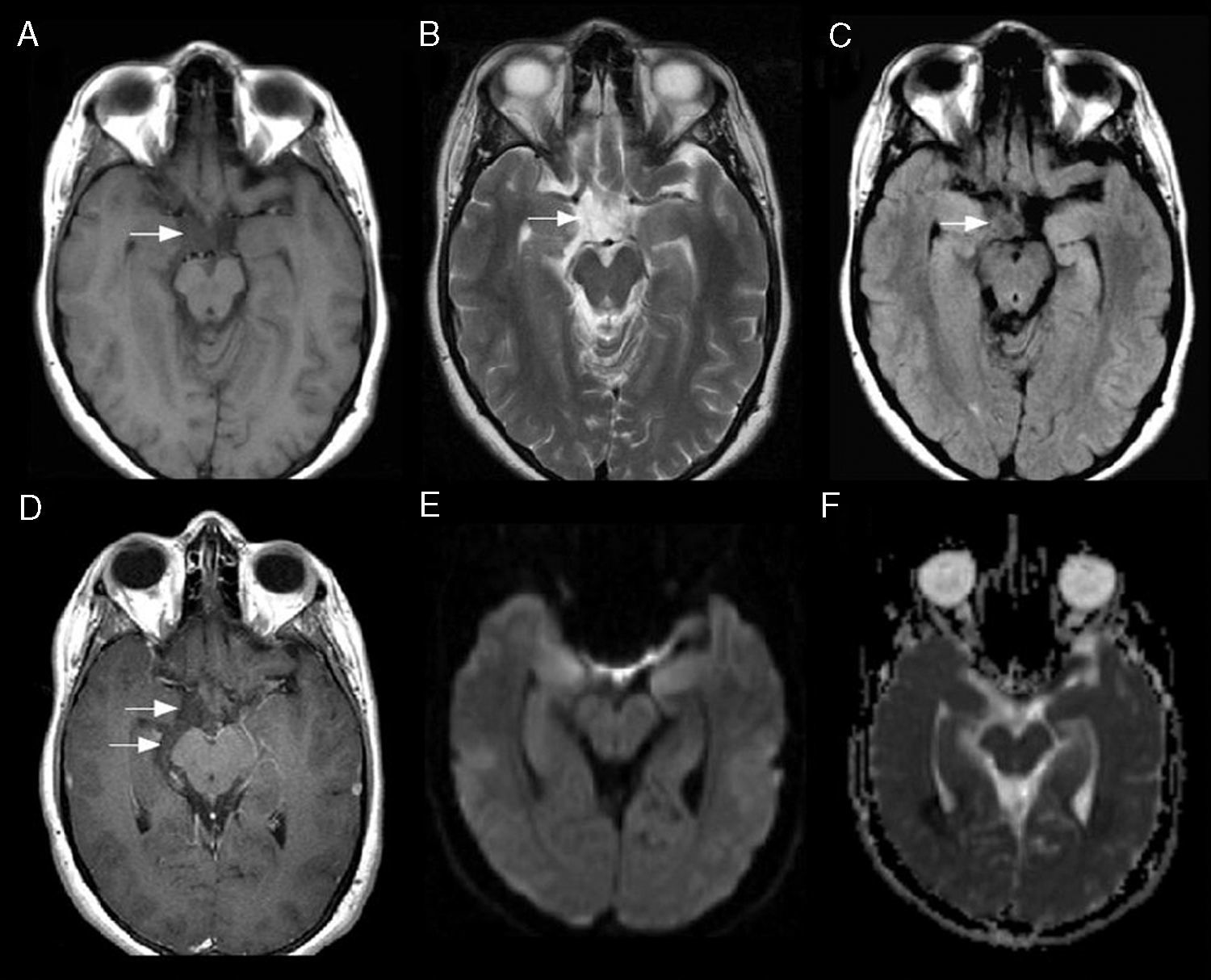
Posteriormente se solicitó resonancia magnética (RM) de cerebro con gadolinio, la cual mostró una lesión localizada en la región supraselar derecha, en relación con la cisterna ambiens e interpeduncular, con leve efecto de masa sobre el nervio óptico y cuerpo mamilar homolateral, siendo hipointensa al parénquima cerebral en T1 y FLAIR, aunque de mayor intensidad de señal que el líquido cefalorraquídeo (LCR). La misma se comportó isointensa al LCR en T2. En difusión no evidenciaba restricción. Tras la administración de contraste endovenoso (gadolinio) no presentó realce (fig. 1).
 y FLAIR (C) e isointensa al LCR en T2 (B), localizada en la cisterna supraselar derecha con extensión a cisternas interpeduncular y ambiens (flechas). La misma tras la administración de contraste endovenoso (gadolinio) no refuerza (D). En difusión (E) y en el mapa de ADC (F) no presenta restricción.")
RM de cerebro, planos axiales: imagen hipointensa al parénquima cerebral en secuencia T1 (A) y FLAIR (C) e isointensa al LCR en T2 (B), localizada en la cisterna supraselar derecha con extensión a cisternas interpeduncular y ambiens (flechas). La misma tras la administración de contraste endovenoso (gadolinio) no refuerza (D). En difusión (E) y en el mapa de ADC (F) no presenta restricción.
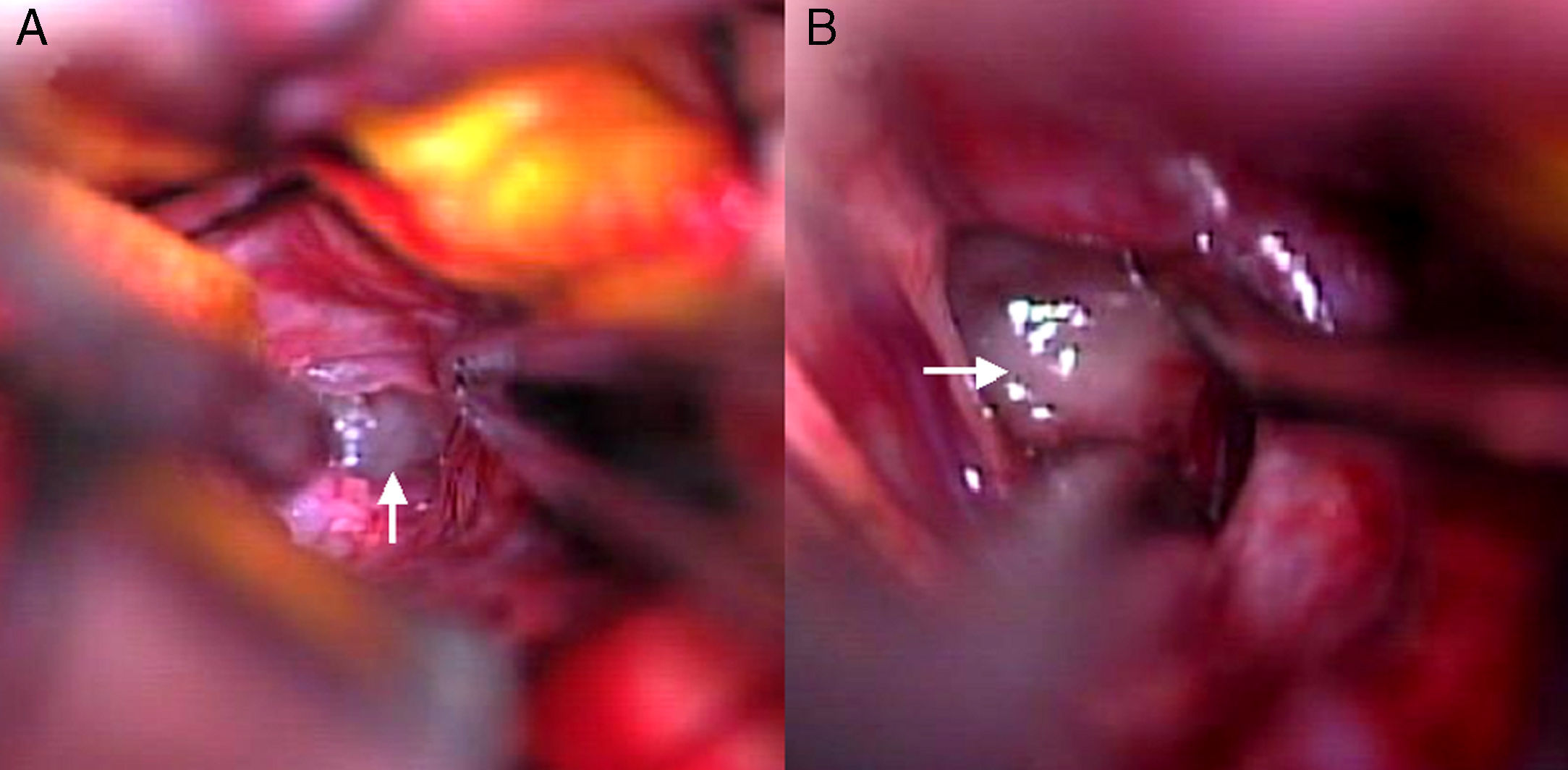
Se realizó interconsulta con el Servicio de Neurocirugía, efectuándose luego biopsia quirúrgica de la masa descrita, donde se visualizó una masa de consistencia gelatinosa y grisácea en la cisterna silviana y supraselar derecha, con extensión al espacio intercarotídeo - óptico homolateral, sin evidencia de afectación parenquimatosa, lo que confirmó su localización extraaxial (fig. 2). Se tomó en forma intraoperatoria una muestra de LCR, la cual no mostró alteraciones.
.")
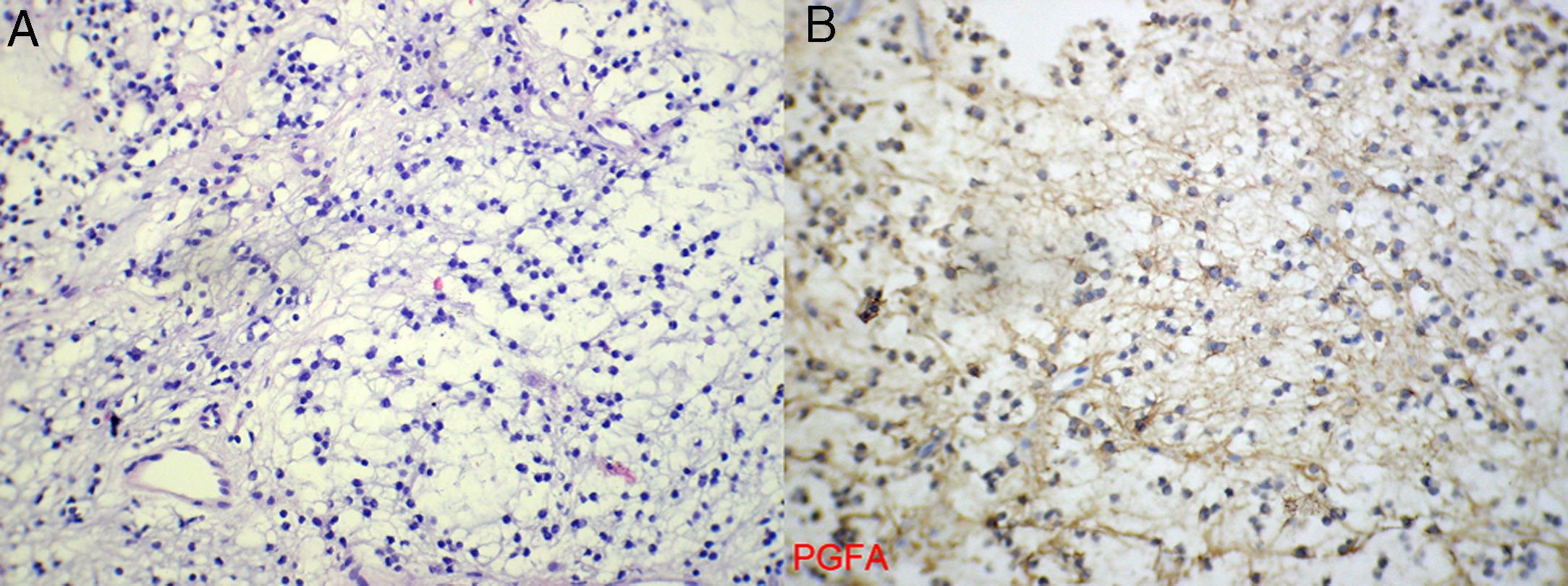
En el análisis de anatomía patológica la pieza quirúrgica estaba compuesta por pequeños fragmentos, algunos laminares, de tejido laxo fibrilar con células pequeñas redondas, en sectores con configuración quística, revestidas por células planas y con un soporte fibroconectivo. El tejido aparentaba ser neuroglial, por lo que se completó con técnicas de inmunohistoquímica (proteína glio-fibrilar ácida positiva). El diagnóstico definitivo fue glioma mixto tipo oligodendro - astrocitoma de bajo grado (fig. 3).
. A. Tejido laxo fibrilar con células pequeñas redondas. B. En la inmunomarcación se constata proteína glio - fibrilar ácida (PGFA) positiva, lo que confirma la naturaleza neuroglial.")
La paciente continúa bajo control neurooncológico e imagenológico, sin cambios en la RM en cuanto a la morfología, intensidad de señal y realce con contraste de la lesión.
ComentariosLa gliomatosis leptomeníngea es una rara condición en la que los tumores crecen en el espacio subaracnoideo, sin una conexión con el parénquima cerebral o la médula espinal. Generalmente es una enfermedad de rápida progresión. La mayoría de estos tumores son astrocíticos de alto grado y el diagnóstico se hace post mortem. La diseminación de oligodendrogliomas en el espacio subaracnoideo (oligodendrogliomatosis leptomeníngea) es generalmente secundaria a la invasión del sistema ventricular o leptomeninges por un oligodendroglioma primario intraparenquimatoso. En contraste con los bien documentados astrocitomas primarios leptomeníngeos, los oligodendrogliomas de este tipo son raros1,2. No encontramos en la literatura ningún caso de oligoastrocitoma primario leptomeníngeo.
Los oligodendrogliomas representan el 2 al 5% de los tumores primarios de cerebro, y el 5 al 18% de todos los gliomas y los oligoastrocitomas mixtos el 9% de todos los gliomas. El pico de incidencia de estos últimos es a los 35 - 45 años, y usualmente se manifiestan con convulsiones3.
La vasta mayoría de los oligodendrogliomas son supratentoriales, siendo el lóbulo frontal el más afectado (50-65%), siguiendo en orden de frecuencia el lóbulo temporal (47%), parietal (7-20%) y occipital (1-4%). Otros sitios incluyen el cerebelo (3%), tronco cerebral y médula espinal (1%), leptomeninges, ángulo pontocerebeloso, ventrículos cerebrales y nervio óptico. La afectación difusa del espacio subaracnoideo y cisternas es característico del oligodendroglioma difuso leptomeníngeo u «oligodendrogliomatosis». Estos ocasionalmente pueden presentar calcificaciones meníngeas3.
Los oligodendrogliomas intraparenquimatosos se manifiestan en la RM como lesiones isointensas al parénquima cerebral en T1 e hiperintensas en T23. No hay características únicas en las neuroimágenes que permitan distinguir un oligodendroglioma de un oligoastrocitoma, aunque se ha visto que estos últimos realzan escasamente con el contraste endovenoso4.
Los tumores primarios leptomeníngeos son raros. Se ha postulado que la presencia de tejido heterotópico glial sería el origen de la gliomatosis leptomeníngea3,5,6. Estas anomalías congénitas consisten en pequeñas islas y nidos de tejido glial dentro del espacio subaracnoideo, las cuales ocurren en aproximadamente el 1% de los individuos sanos y en el 25% de los pacientes con otras malformaciones cerebrales7–10. La transformación neoplásica del tejido glial heterotópico es un evento raro, generalmente diagnosticado en la autopsia11. La mayoría de los casos reportados de gliomas primarios leptomeníngeos son astrocitomas o glioblastomas, los cuales pueden ser bien localizados y pasibles de resección quirúrgica, o difusos, irresecables y eventualmente fatales12.
El diagnóstico premortem de la gliomatosis difusa leptomeníngea es difícil debido a los síntomas neurológicos inespecíficos, el curso clínico fluctuante y el examen negativo del LCR para células neoplásicas9.
No es sorpresivo que los criterios para la gliomatosis leptomeníngea no estén firmemente establecidos. Esta situación se debe en gran parte a la necesidad de excluir el diagnóstico más probable de diseminación leptomeníngea de un glioma intraparenquimatoso. Se han propuesto tres criterios necesarios antes del diagnóstico de gliomatosis leptomeníngea: 1) ausencia de compromiso del parénquima cerebral o medular subyacente por el tumor extraaxial; 2) sin evidencia de un tumor primario en el neuroeje; y 3) existencia de una cápsula alrededor del tumor13.
En la histopatología la mayoría de los gliomas de bajo grado son tumores no encapsulados que en general infiltran en forma difusa. Son masas sólidas y ligeramente grisáceas, cuya consistencia varía desde blanda hasta casi gelatinosa, como en el caso de nuestra paciente (fig. 2). Puede haber degeneración quística. Microscópicamente se caracterizan por una proliferación de células gliales fibrilares bien diferenciadas, con pocas mitosis y densidad celular baja o moderadamente elevada. La mayoría muestra una intensa inmunorreactividad a anticuerpos frente a la proteína ácida fibrilar glial14 (fig. 3).
Dentro de los diagnósticos diferenciales de las lesiones extraaxiales a nivel supraselar, como en el caso de nuestra paciente, se incluye el tumor epidermoide. Se trata de un tumor congénito benigno que contiene detritus, queratina y colesterol. En secuencia T1 de RM presenta típicamente señal intermedia y no realza con contraste endovenoso. En T2 es hiperintenso, similar al LCR. En difusión presenta alta intensidad de señal, diferenciándose fácilmente del LCR adyacente o quistes aracnoideos. Esa es la clave imagenológica que nos orienta hacia otra etiología probable de la masa supraselar15–17. Los diagnósticos diferenciales de la gliomatosis difusa leptomeníngea incluyen diseminación leptomeníngea de un tumor primario intracerebral, histiocitosis, meningitis infecciosa y paquimeningitis idiopática. Clínicamente la gliomatosis leptomeníngea produce un cuadro similar a la meningitis crónica infecciosa, particularmente tuberculosa o fúngica, en las cuales el análisis del LCR es generalmente negativo18,19. La apariencia en la TC o en la RM del realce leptomeníngeo difuso, con o sin hidrocefalia, es difícil de distinguir de una forma subaguda de meningitis, de etiología infecciosa o no20.
En el caso de nuestra paciente la afectación leptomeníngea no fue difusa como en la mayoría de los casos reportados, sino que estaba confinada al espacio subaracnoideo de la cisterna supraselar, confirmado en la Resonancia Magnética, en forma intraoperatoria así como en el análisis del LCR. Además, el realce poscontraste no fue intenso, probablemente debido a que el tumor era de bajo grado. Estas características en las imágenes excluían en un primer momento como diagnóstico probable un proceso tumoral agresivo o causas infecciosas.
En última instancia el diagnóstico definitivo en este tipo de lesiones se basa en la obtención de una biopsia y su confirmación histológica1.
