




El síndrome de Leigh o encefalomielopatía necrotizante subaguda es una enfermedad neurodegenerativa progresiva, genética y clínicamente heterogénea que se asocia por lo general con alteración del metabolismo mitocondrial a nivel de la fosforilación oxidativa. Se presenta generalmente en el periodo neonatal, pero también en la edad adulta, en la que suele expresarse con una combinación de diversas manifestaciones entre ellas parkinsonismo, alteraciones del movimiento, alteraciones neuropsiquiátricas y otras.
Caso clínicoPaciente masculino de 34 años de edad, que inició sus síntomas a los 18 años, algunos meses después de que su madre falleciera con síntomas parecidos, pero sin diagnóstico definido. El paciente presentó mioclonías progresivas en miembro superior derecho, espasmos palpebrales bilaterales y cervicales, asociándose posteriormente, mioclonia facial desencadenado por el habla y distonía en miembro inferior derecho que le impedía la deambulación. La resonancia magnética de encéfalo mostró la presencia de lesiones simétricas bilaterales en núcleos basales, lesiones corticales y periacueductales. El estudio genético detectó la mutación m.14487T>C en el gen ND6 del complejo 1 del sistema de fosforilación oxidativa mitocondrial en los leucocitos y células de mucosa bucal. El estudio se extendió a la abuela y dos tías maternas, asintomáticas en ese momento, quienes portaban la misma mutación.
ConclusiónEl caso corresponde a un paciente adulto, con síndrome de Leigh, portador de la mutación m.14487T>C, la cual parece estar relacionada con un fenotipo predominantemente mioclónico progresivo, de origen cortical.
Leigh's syndrome or subacute necrotizing encephalomyelopathy, is a progressive, genetically and clinically heterogeneous neurodegenerative disease that is generally associated with alteration of mitochondrial metabolism at the level of oxidative phosphorylation. It generally occurs in the neonatal period but also in adulthood, at this age it is usually expressed with a combination of various manifestations including Parkinsonism, movement disorders, neuropsychiatric disorders and others.
Clinical caseThirty four-year-old male patient who began his symptoms at 18 years of age, after a few months his mother died with similar symptoms, but without a defined diagnosis. The patient presented progressive myoclonus in the right upper limb, bilateral palpebral and cervical spasms, later associated with facial myoclonus triggered by speech and dystonia in the lower right limb that restrict him from ambulation. Brain MRI showed the presence of bilateral symmetric lesions in the basal ganglia, cortical and periaqueductal lesions. The genetic study showed a m.14487T>C mutation in the ND6 gene of complex 1 of the mitochondrial oxidative phosphorylation system in leukocytes and cells of the buccal mucosa. This study was extended to the grandmother and two maternal aunts, asymptomatic at the time, who carried the same mutation.
ConclusionThe case corresponds to an adult patient with Leigh syndrome, a carrier of the m.14487T>C mutation, which seems to be related to a predominantly progressive myoclonic phenotype of cortical origin.
El síndrome de Leigh (SL) o encefalomielopatía necrotizante subaguda1 es un trastorno genético que produce disfunción mitocondrial por mutaciones que pueden originarse en el genoma nuclear o el genoma mitocondrial. El patrón de herencia puede ser autosómico recesivo, ligado al cromosoma X o esporádico. Dichas mutaciones generan alteraciones en el funcionamiento de la cadena respiratoria, siendo las del complejo I las más frecuentes1,2.
El SL suele presentarse en 80% de los casos en los primeros meses de vida, con un curso severo, rápidamente progresivo y en el cual los pacientes suelen fallecer antes del año de vida; estos casos se suelen vincular a mutaciones del genoma nuclear3. Sin embargo, 20% de los pacientes inician la enfermedad luego del primer año y hasta la adultez con un curso lentamente progresivo, y vinculado a mutaciones del ADN mitocondrial4. Las manifestaciones clínicas características son parkinsonismo, distonias, mioclonías, alteración neuropsiquiátrica, nistagmus, oftalmoparesia, ataxia, atrofia óptica y otras, sin embargo, sus manifestaciones específicas van a depender de la mutación causante y el grado de heteroplasmia en los diferentes tejidos afectados3,4.
Presentamos el caso de un paciente masculino con un cuadro predominantemente mioclónico y rígido distónico progresivo de inicio tardío en el que se identificó una mutación mitocondrial relacionada con el SL.
Caso clínicoMasculino de 34 años de edad procedente de Lima, soltero, hijo único, cuya madre falleció con un cuadro clínico similar, pero sin diagnóstico precisado. Inició a los 18 años de edad con mioclonías en mano derecha desencadenados por la escritura y la realización de movimientos finos, que posteriormente también comprometió al brazo derecho. Estos episodios calmaban completamente con la ingesta de clonazepam 0,5 mg 1 tableta cada 24 horas. Un año después se añadieron contracciones involuntarias de musculatura escapular que desplazaban el miembro superior derecho hacia atrás, por lo que se aumentó dosis de clonazepam a 1 mg/día, con lo que las contracciones remitieron completamente.
Dos años más tarde se agregaron mioclonías palpebrales episódicas que duraban aproximadamente 20 minutos, por lo que es referido a hospital psiquiátrico por sospecha de trastorno conversivo.
A los 25 años se manifestaron contracciones episódicas involuntarias de musculatura faríngea, desencadenadas por el habla que le dificultaban articular palabras y le generaban emisión de sonidos agudos, sin compromiso de la deglución. Desde ese tiempo presentó disartria y voz nasal progresiva, realizándosele resonancia magnética de encéfalo que en ese momento resultó normal.
Cuatro meses antes de la hospitalización presentó parestesias en miembro inferior derecho y contracciones episódicas, en el muslo y abdomen inferior ipsilateral, que mantenían dicho miembro en extensión y dificultaban la deambulación. Un mes después se agregaron mioclonías desencadenadas por el habla en hemicara y región cervical izquierda que dificultaban la articulación de palabras y se aliviaban parcialmente al presionar la musculatura cervical izquierda, lo cual además le permitía mejorar la articulación de palabra, siendo referido para reevaluación neurológica.
Al examen neurológico el paciente se encontraba vigil, orientado en tres esferas, con funciones mentales superiores y fuerza muscular conservada. Se observaron mioclonías palpebrales, en hemicara y regiones cervicales izquierdas desencadenadas por el habla asociado a disartria. Presentó mioclonías en región abdominal y de miembros inferiores que le dificultaban la marcha y acciones como levantarse de la cama. Presentaba además, hipertonía en miembro inferior derecho, hiperreflexia generalizada, clonus derecho, dismetría e hipotonía izquierda. No mostró trastorno deglutorio.
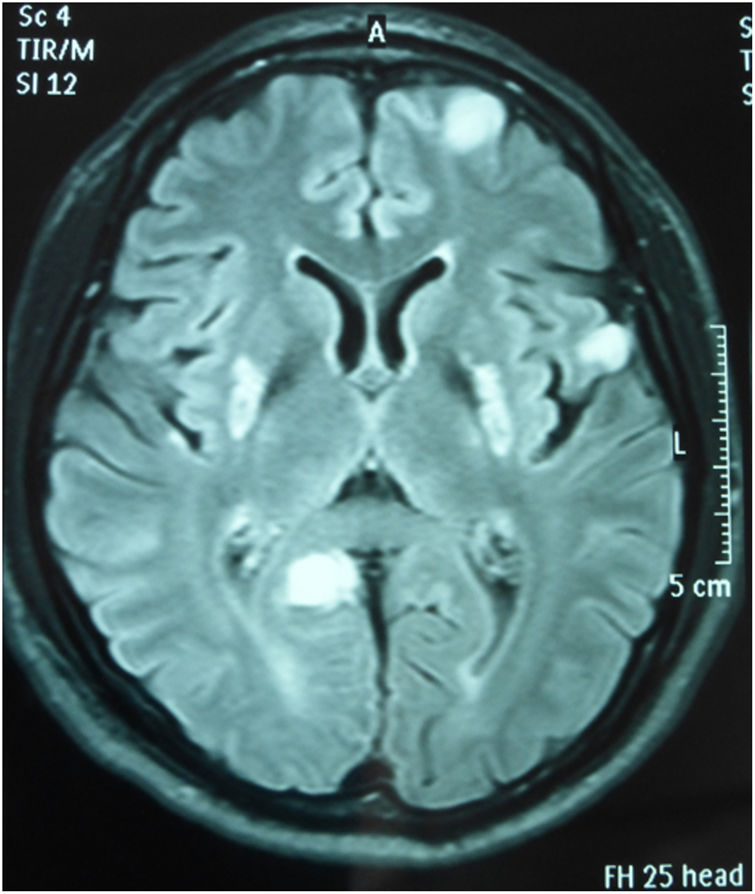
Los exámenes hematológicos de ingreso (hemograma, glucosa, urea, creatinina, ceruloplasmina, cobre, tiamina y ácido láctico) fueron normales. El electroencefalograma mostró disfunción cortical frontal bilateral epileptogénica y en la resonancia magnética de encéfalo, se evidenció señal hiperintensa en la secuencia FLAIR en los núcleos lenticulares en forma simétrica además de compromiso de sustancia gris periacueductal y núcleos rojos, así como múltiples lesiones corticales en regiones frontal y temporal derecha, y occipital izquierda (fig. 1).

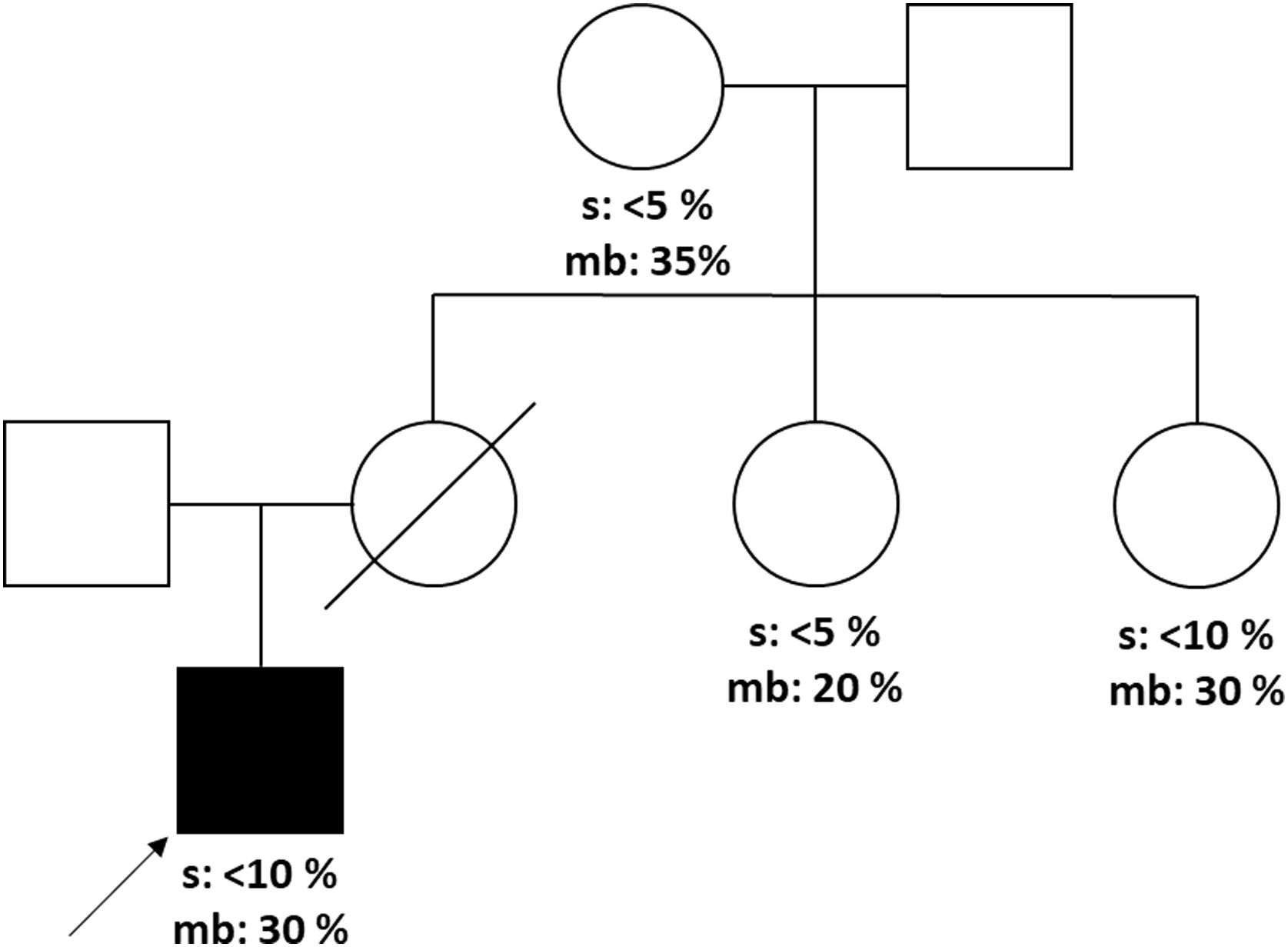
El estudio genético mitocondrial del paciente (caso índice) permitió identificar la mutación m.14487T>C con un porcentaje de heteroplasmia menor al 10% en células sanguíneas y de 30% en células de mucosa bucal. Los familiares relacionados por vía materna (abuela y dos tías) estudiados, en ese momento asintomáticas también presentaron la mutación en ambos tejidos con diferentes valores de heteroplasmia (fig. 2).

El paciente recibió carbamazepina 700 mg/día, levetiracetam 500 m/día, clonazepam 5 mg/día presentando un adecuado control de las crisis mioclónicas en hemicara izquierda y la hipertonía en miembro inferior derecho.
DiscusiónEl paciente inició sus síntomas a los 18 años, con un cuadro mioclónico lentamente progresivo, inicialmente focal, que comprometía miembro superior derecho. Estos síntomas se asociaron a un cuadro rígido distónico, y trastorno de la marcha que llevó a la postración. La resonancia magnética de encéfalo, mostró la presencia de lesiones bilaterales y simétricas que comprometían los núcleos basales, sustancia gris periacueductal, núcleos rojos y regiones frontales y temporales corticales. El examen genético demostró la presencia de una mutación mitocondrial, con lo cual, previo descarte de otras causas de lesión estriatal bilateral, se realizó el diagnóstico de SL de inicio tardío.
El paciente cumplió con los criterios clínicos, imagenológicos y genéticos propuestos por Baertling et al.,1 para la definición del SL. La resonancia magnética de encéfalo fue el principal elemento de sospecha inicial de la enfermedad, ya que mostró todas las características consideradas como típicas para SL. La ausencia de afectación del cuerpo mamilar, y la presencia de tiamina sérica en valores normales, alejó la posibilidad de enfermedad de Wernicke.
El antecedente de enfermedad materna sugirió un tipo de transmisión mitocondrial. A pesar de que no se realizó la cuantificación de la actividad de los complejos de la cadena respiratoria mediante biopsia, y que el lactato en sangre y en líquido cefalorraquídeo (LCR) fue normal, se identificó la mutación m.14487T>C, la cual causa la sustitución del aminoácido metionina por valina en la posición 63, en la región transmembrana más conservada de la subunidad ND6 generando disfunción del complejo I de la cadena respiratoria mitocondrial5,6.
Esta inusual mutación, que por primera vez se describe en Perú, se asocia a SL o necrosis estriatal bilateral, solo en 0,3% de los casos, con niveles variables de heteroplasmia (65-95%)5.
La heteroplasmia es una característica propia de las mutaciones mitocondriales y hace referencia al diferente grado en que la mutación se expresa en los tejidos, y que puede influir en el cuadro clínico. Se ha observado que valores altos de heteroplasmia de la mutación m.14487T>C en las células sanguíneas se relacionaría con presentaciones tempranas y agresivas de la enfermedad y que porcentajes menos severos se relacionarían con las formas tardías y de evolución más lenta7. Nuestro paciente presentó niveles relativamente bajos de heteroplasmia, en células sanguíneas y de mucosa bucal, lo cual explicaría la presentación tardía y progresiva de este síndrome en este caso, los familiares por vía materna del paciente (abuela y tías) también presentaron niveles bajos de heteroplasmia, pero similares e incluso mayores en comparación con el paciente índice, sin embargo a diferencia de este, eran asintomáticas, este hallazgo aparentemente contradictorio podría explicarse porque las células estudiadas no fueron las del sistema nervioso, sustrato histopatológico de los síntomas del paciente, en las cuales posiblemente se hubiera observado un mayor porcentaje de heteroplasmia y una mejor correlación clínico molecular.
La mutación m.14487T>C da lugar a una amplia variabilidad de fenotipos, según su porcentaje de heteroplasmia, incluyendo el SL, encefalomielopatía mitocondrial con acidosis láctica y episodios de stroke like (MELAS) y neuropatía óptica de Leber (LHON), e incluso manifestaciones inespecíficas como, epilepsia, migraña o hipoacusia neurosensorial7-9.
Sin embargo, dentro de esa variedad de manifestaciones, algunos autores como Dermaut et al7. y Raspall-Chaure et al.8, han coincidido en describir como características propias de la mutación m.14487T>C, la presencia de un cuadro mioclónico progresivo, asociado a un síndrome extrapiramidal en forma de distonía y síndrome hipoquinético rígido progresivo. Este perfil clínico coincide con el de nuestro paciente, en el que además se logró demostrar, al igual que en los casos de Dermaut et al. y Raspall-Chaure et al., el diagnóstico de epilepsia mioclónica progresiva, sustentado en la presencia de grafoelementos epileptiformes en el electroencefalograma.
La presencia de mioclonías faciales como característica predominate del SL asociado a esta mutación, fue comunicado previamente por Navarro et al.,10 quienes reportaron un paciente que presentaba mioclonías faciales desencadenadas por la masticación. Nuestro paciente presentaba también mioclonías faciales, pero desencadenadas por el habla, lo que sugiere que esta mutación se relacionaría con un fenotipo adulto particular caracterizado por la presencia de mioclonías progresivas, que probablemente comprometan la región facial y se desencadenen por estímulos motores como la masticación y el habla.
En conclusión, el SL debe considerarse en pacientes adultos que se presenten con un síndrome neurológico progresivo predominantemente mioclónico y rígido distónico y que además presenten una imagen en resonancia magnética compatible, en estos casos la mutación m.14487T>C debe ser evaluada especialmente si las mioclonías comprometen la región facial.
Responsabilidades éticasTodas las investigaciones se han regido por los principios de la Declaración de Helsinki y la metodología de genética-molecular fue aprobada por el Comité de Ética (CEICA) N° 13/2017.
Conflicto de interesesLos autores declaran no tener conflicto de intereses.
Los trabajos aquí descritos han sido subvencionados por el Fondo de Investigaciones Sanitarias (PI17-00021; PI17-00166); Programa de crowdfunding Precipita-FECYT (PR194); Gobierno de Aragón (Grupos Consolidados B33_17R) y FEDER 2014-2020 «Construyendo Europa desde Aragón»; Asociación de Enfermos de Patología Mitocondrial (AEPMI); CIBERER es una iniciativa del Instituto de Salud Carlos III.
