




El diagnóstico y el tratamiento de las malformaciones congénitas todavía hoy constituyen una problemática universal. El presente trabajo corresponde al estudio de un caso de un lactante de 6 meses de edad, paciente del Hospital Oncológico Dr. Julio Villacreses Colmont de Manabí (SOLCA), a partir del mes de mayo del 2015, con cuadro clínico de aumento de la circunferencia cefálica, babeo y en ocasiones vómitos, irritabilidad y retraso del desarrollo psicomotor, rostro inexpresivo, sin tratamiento específico. Una vez realizada la valoración clínica del paciente, con el apoyo de las pruebas diagnósticos de imagenología(ecografía abdominopélvica y resonancia magnética cerebral) se pudieron identificar múltiples malformaciones congénitas, entre ellas se observó a parte de las neurológicas la agenesia del renal izquierda, lo cual es raro en esta morbilidad. El objetivo de este trabajo consiste en demostrar la elevada frecuencia de esta morbilidad. En Ecuador no reportan estadísticas del síndrome o existe un subregistro de esta entidad.
The diagnosis and treatment of congenital malformations continues to be a universal problem. This work is a study of a case of a six-month-old infant patient treated at the Cancer Hospital Dr. Julio Villacreses Colmont Manabi (SOLCA) from May 2015, with clinical symptoms of increased cephalic girth, drooling and occasional vomiting, irritability and delayed psychomotor development, expressionless face, without specific treatment. We conducted clinical assessment, with abdominopelvic ultrasound and brain MRI, and found multiple congenital malformations, including Moebius syndrome–a rare disease. The aim of this paper is to demonstrate the high frequency of this disease. In Ecuador, there are no statistics of this syndrome, or else, the disease is underreported.
Las estructuras encefálicas aparecen luego de ocurridos 4 procesos básicos: a) proliferación neuronal; b) migración; c) período de organización, el cual se establece la diferenciación celular; este se desarrolla hasta el nacimiento una vez establecido el patrón de funcionamiento de las diferentes regiones encefálicas, y d) mielinización. Durante la cuarta semana, después del cierre de los neuroporos, el extremo cefálico del tubo neural craneal al cuarto par de somitos se dilata considerablemente y aparecen las 3 vesículas encefálicas primarias a partir de las cuales se origina el encéfalo: prosencéfalo (cerebro anterior), mesencéfalo (cerebro medio) y rombencéfalo (cerebro posterior). Simultáneamente, se están formando 2 plegamientos: el pliegue cervical, en la unión del rombencéfalo, y médula espinal y el pliegue cefálico en el mesencéfalo. El mesencéfalo limita con el rombencéfalo por un surco: el istmo del rombencéfalo (de His). Durante la quinta semana, el prosencéfalo y el rombencéfalo se dividen en 2 vesículas secundarias: el prosencéfalo origina el telencéfalo (cerebro terminal), que consta de una parte media y 2 evaginaciones laterales (hemisferios cerebrales primitivos), y el diencéfalo, que presenta la evaginación de las vesículas ópticas. El rombencéfalo formará finalmente el metencéfalo, que constituirá la protuberancia y el cerebelo el mielencéfalo, el futuro bulbo raquídeo. El límite metencéfalo-mielencéfalo queda definido por el pliegue protuberancial. Este pliegue se origina debido al crecimiento desigual del rombencéfalo, dando lugar a un adelgazamiento de su techo. Al principio, el encéfalo tiene su estructura básica muy similar a la médula espinal; sin embargo, debido a la aparición de los pliegues y surcos encefálicos, se producen variaciones considerables en la disposición de los diferentes elementos. En general, las placas alares y basales del rombencéfalo y mesencéfalo se encuentran bien definidas; en cambio, en el prosencéfalo las placas alares están acentuadas y las basales en plena regresión.
El rombencéfalo está formado por: mielencéfalo y metencéfalo1.Mielencéfalo: corresponde a la vesícula encefálica más caudal y se diferencia en el bulbo raquídeo (médula oblonga). Sus paredes laterales presentan cierta eversión, tal como se abren las conchas de una almeja; sin embargo, su estructura general es bastante parecida a la de la médula espinal. Los neuroblastos de las placas alares migran a la capa marginal en dirección ventrolateral para formar los núcleos olivares. Ventralmente, las fibras corticoespinales que descienden desde la corteza cerebral (giro precentral) forman las denominadas pirámides. El pliegue protuberancial hace que las paredes bulbares laterales se desplacen lateralmente y que la placa del techo se extienda y adelgace considerablemente. Como consecuencia, la cavidad del mielencéfalo (futuro iv ventrículo) toma forma romboide y los núcleos motores pasan a ubicarse medialmente a los núcleos sensitivos. Las placas alares y basales están bien definidas. La placa basal contiene 3 grupos de núcleos motores: eferente somático o medial, eferente visceral especial o intermedio y eferente visceral general o lateral. Estos 3 grupos originan los núcleos motores de los nervios craneales ix, x, xi y xii, que se ubican en el piso del cuarto ventrículo medial al surco limitante. La placa alar contiene 3 grupos nucleares sensitivos: aferente somático o lateral, aferente visceral especial o intermedio y aferente visceral general o medial. Estos grupos neuronales forman los núcleos sensitivos de los nervios craneales v, vii, viii, ix y x, y los núcleos graciles y cuneatus1.
Embriológicamente, cuando este proceso presenta alguna alteración, y no ocurre el desarrollo de las estructuras anatómicas, se producen varias alteraciones neurológicas conocidas en la terminología médica como anomalías congénitas, una de ellas es el síndrome de Moebius (MBS), el cuadro clínico de esta morbilidad.
Reporte del casoLa presentación del caso, con los respectivos resultados de las pruebas diagnósticas y su confirmación, se realizó con previa autorización de los padres mediante el consentimiento informado.
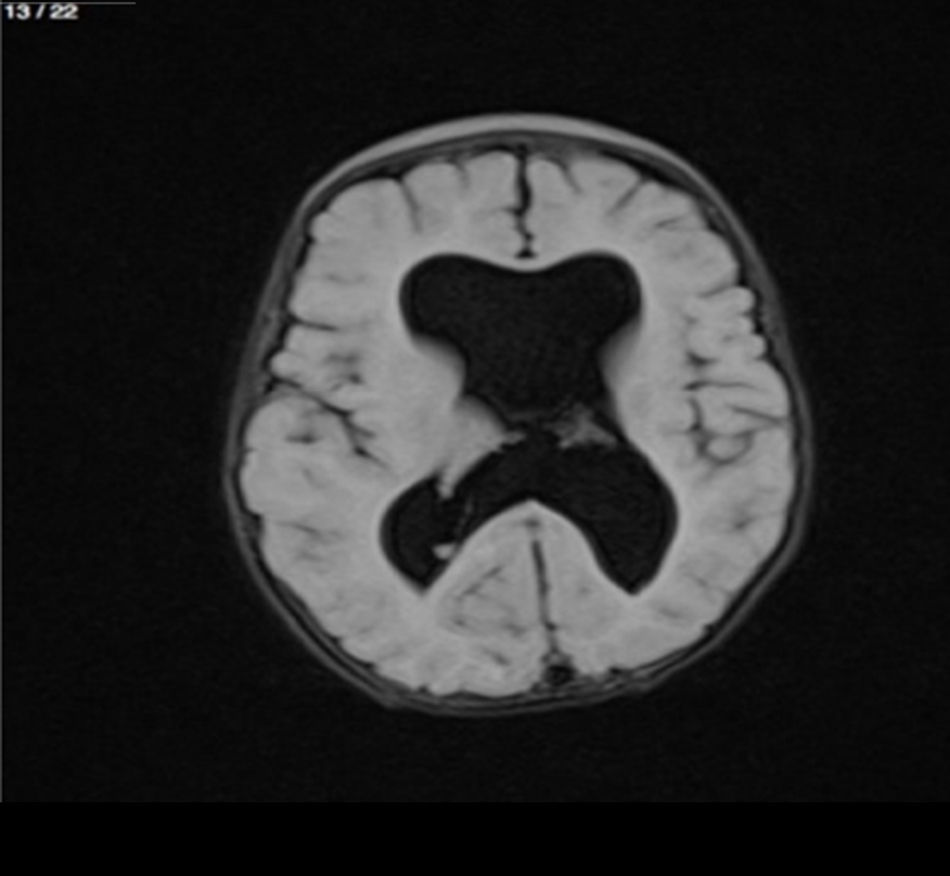
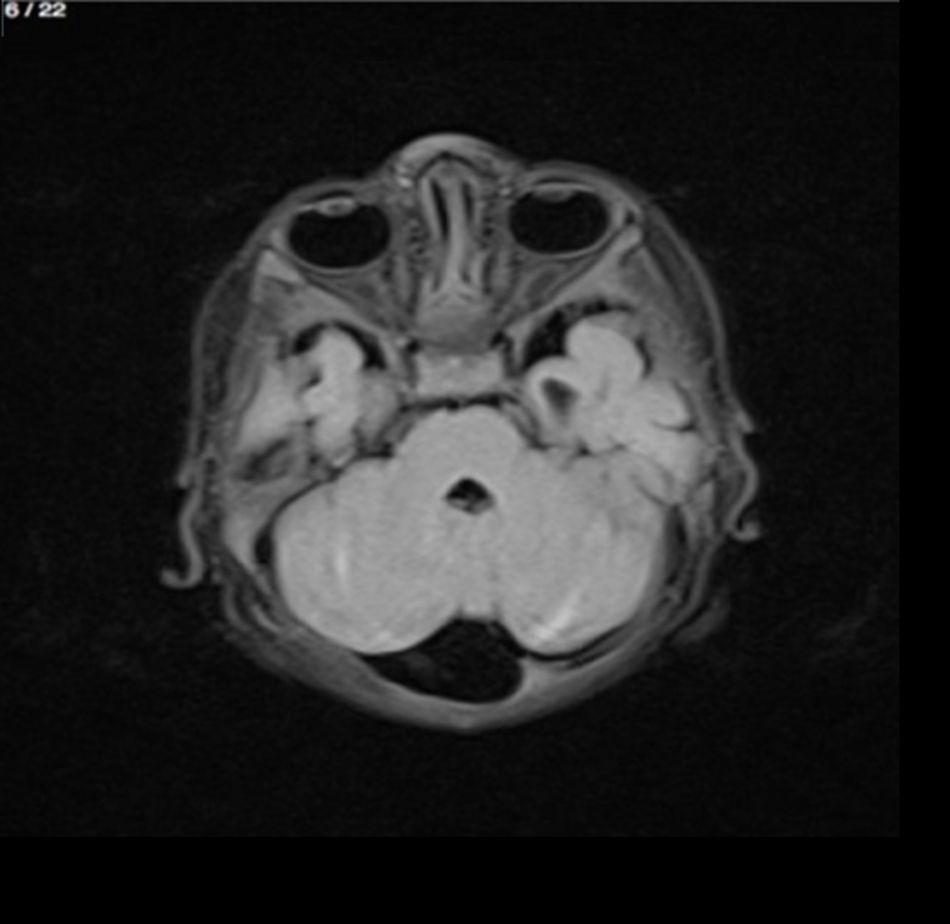
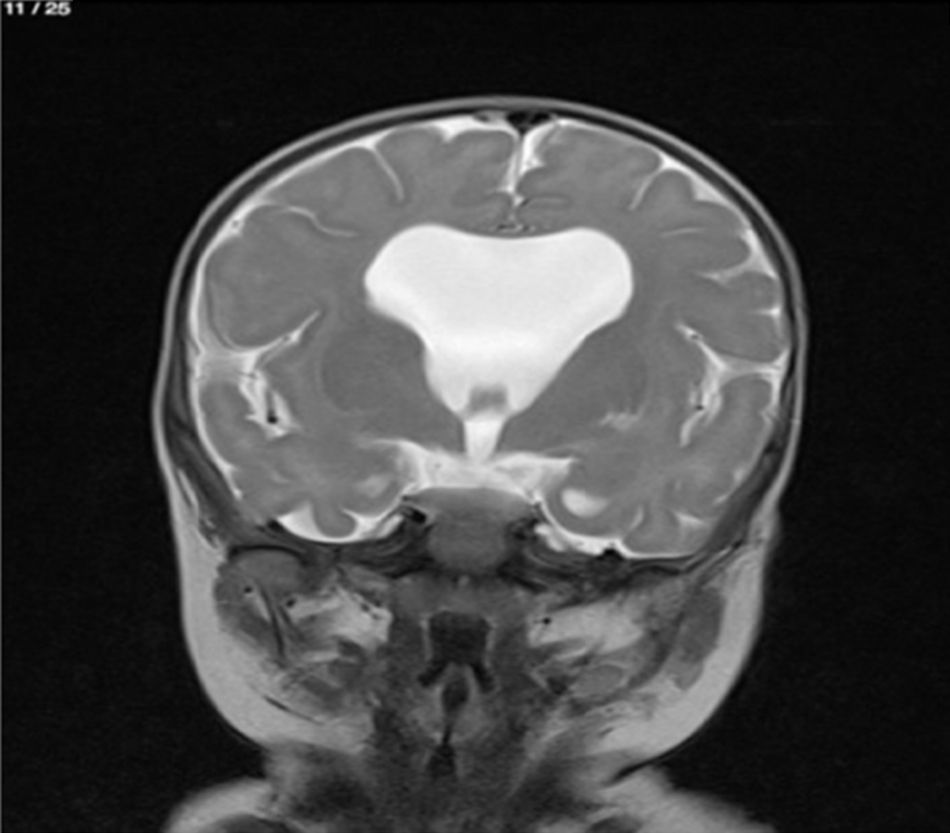
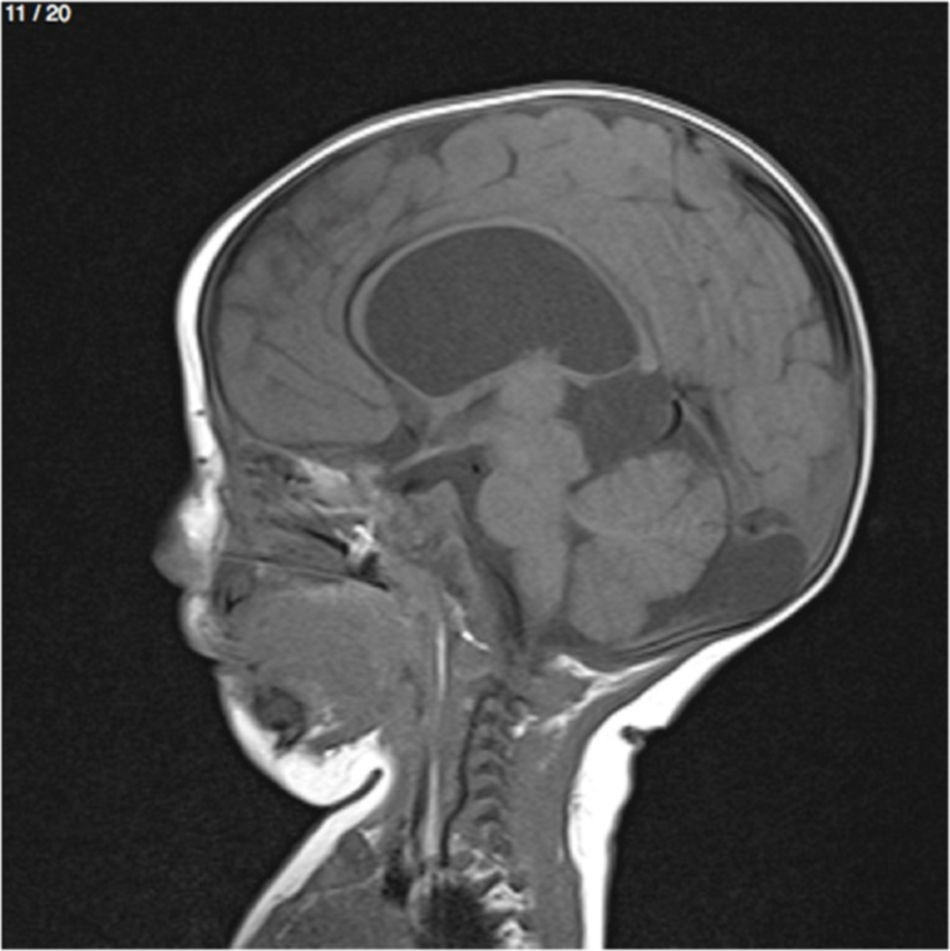
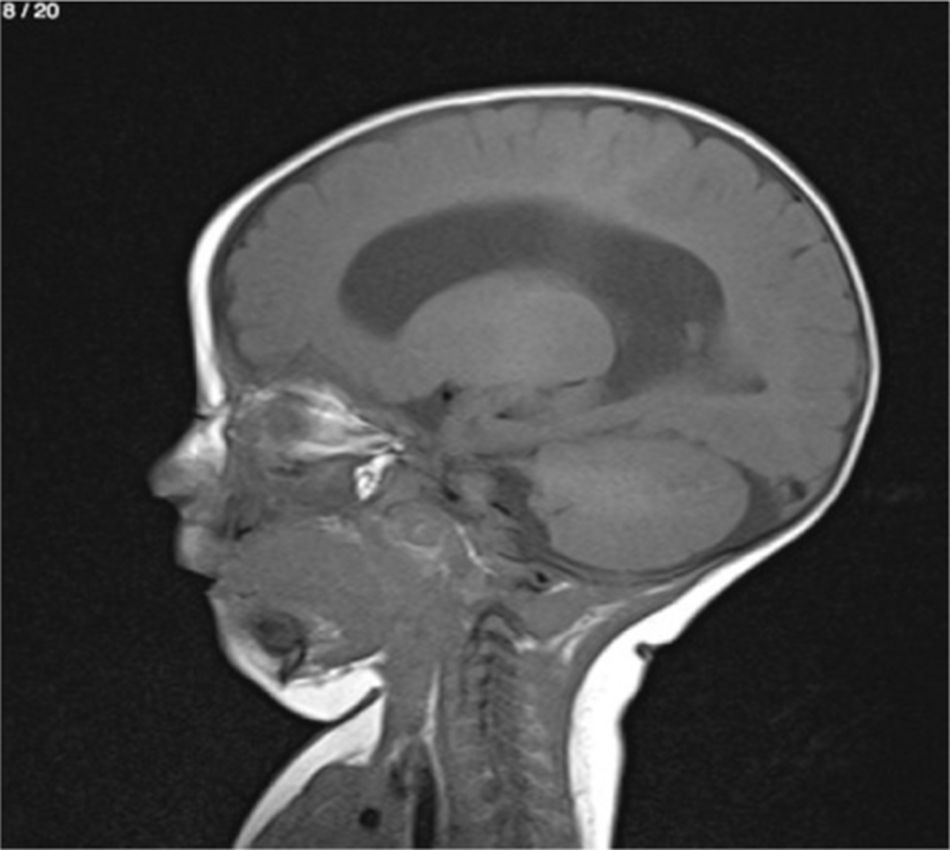
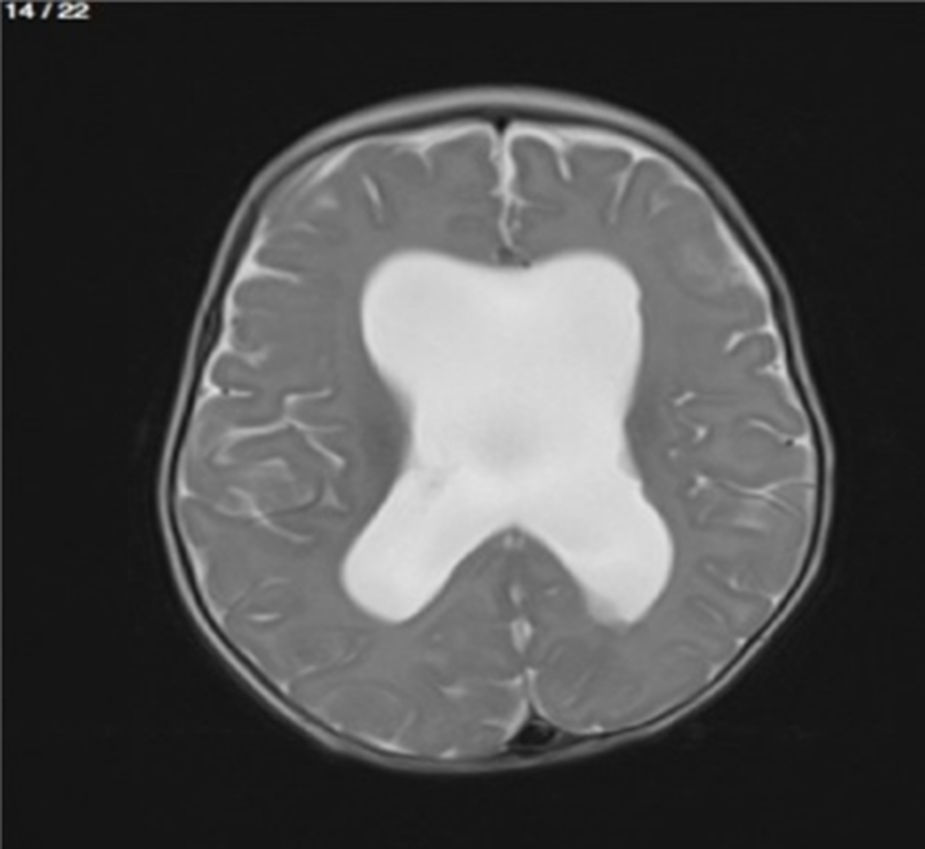
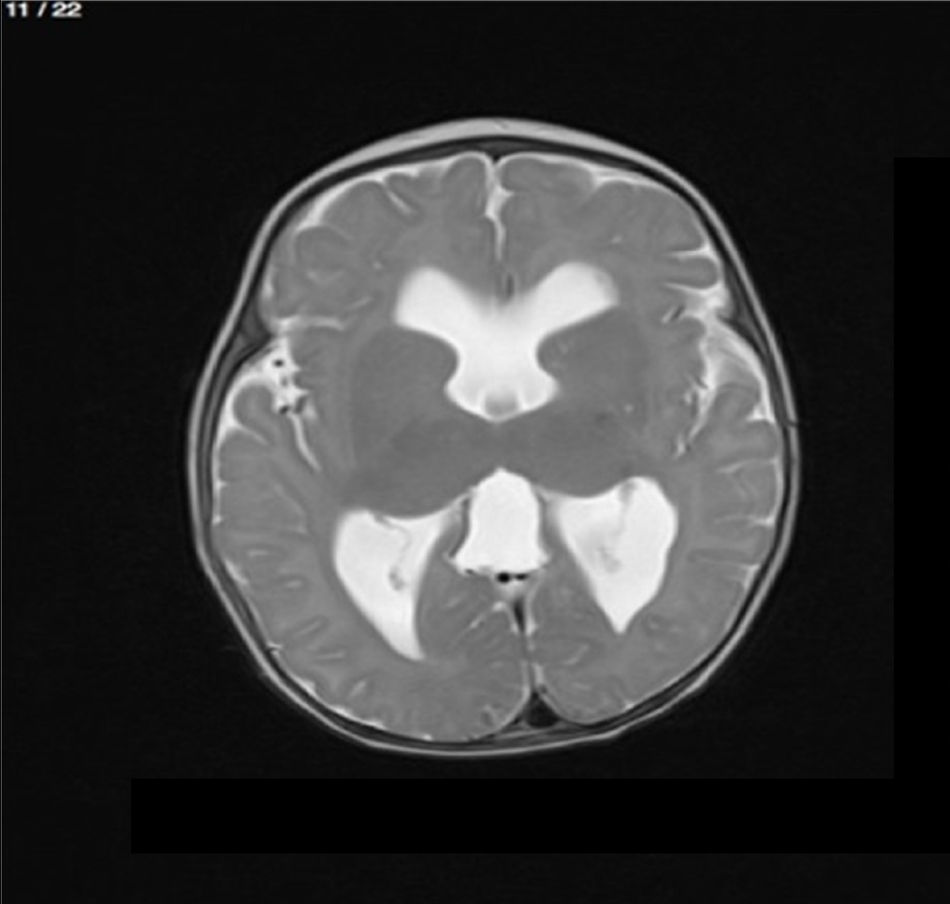
Lactante MSBE, de 6 meses de edad, ecuatoriano, de piel blanca, con antecedentes de retardo del desarrollo psicomotor, con aumento del perímetro cefálico 6cm por encima de lo normal, frente, fontanela bregamática amplia de 6LX5A con trastorno neurológico periférico visible; acude a consulta por presentar aumento de la circunferencia cefálica, babeo, en ocasiones vómitos, irritabilidad, retraso del desarrollo psicomotor; no sostiene la cabeza, no respuesta al extraño y vómitos frecuentes. En función de este cuadro clínico, se solicita una resonancia magnética de cerebro sin contraste y una ecografía abdominopélvica para la búsqueda de más alteraciones orgánicas (figs. 1-7).

.")





El MBS es considerada una enfermedad rara, descrita en 1880 por von Graefe y Saemish, quienes en esa época agruparon a algunos pacientes con trastornos congénitos infrecuentes de la región facial y no progresivos, lo cual fue validado más tarde, en 1888, por Paul Julius Moebius, con el nombre de «síndrome de Moebius», definido como «parálisis congénita de los núcleos de los pares craneales vi motor ocular externo, vii facial y otros pares craneales como: xii hipogloso, iii motor ocular común, cuyo espectro clínico es variable; afecta a otros pares craneales (v, x, xi y xii) y se asocia con múltiples malformaciones, entre ellas musculoesqueléticas; el retraso mental y el autismo se han reportado en algunos casos2-4.
Estas malformaciones anatómicamente se caracterizan por un desarrollo incompleto o parcial de 2 pares craneales que dan lugar a los movimientos oculares y la mímica facial, son el vi y el vii par craneal, respectivamente; en un estudio realizado en México para detallar los hallazgos oftalmológicos más importantes se encontraron que el mayor porcentaje lo tenía la limitación ojo secuestro (100%), la parálisis facial (100%), esotropía (54%), epicanto (51,5%), entropión (22%) y la historia de uso de inductores de aborto en la madre en el primer trimestre del embarazo (28%). También se encontraron hipertropía y exotropía en algunos casos, llegando a la conclusión de que el MBS tiene un amplio espectro de manifestaciones oftalmológicas que son importantes para detectar temprano con el fin de mejorar la función y la estética5.
Pueden estar comprometidos otros puntos cerebrales y afectar a funciones sensoriales de relevancia clínica como son el xii par craneal (hipogloso), que da lugar a dificultades iniciales para tragar que pueden llevar a déficit de desarrollo de la lengua y los problemas que conllevan la falta de sonrisa, el babeo, el habla y la pronunciación defectuosas, además del compromiso del iii par craneal (motor ocular común), cuyos principales signos son el estrabismo y la limitación del movimiento del globo ocular, movimientos del espejo «son un trastorno guía axonal que consiste en movimientos involuntarios contralaterales que imitan los intencionales unilaterales, que implican normalmente los dedos de la mano, los síndromes de Klippel-Feil, síndrome de Kallman o hemiplejía congénita, la mutación autosómica dominante en los genes DCC o RAD51 y el síndrome de retracción de Duane guardan relación con esta morbilidad; de la misma forma, existen trabajos en los cuales se han encontrado malformaciones cardiacas, como la de Taussing Bing, que consiste en una doble salida ventricular derecha (DSVD) en la que la comunicación interventricular se encuentra debajo de la arteria pulmonar. Este tipo de DSVD también se denomina anomalía de Taussig-Bing. Con este tipo de DSVD, la sangre rica en oxígeno sale del ventrículo izquierdo a través de la comunicación interventricular y llega a la arteria pulmonar. La sangre pobre en oxígeno del ventrículo derecho fluye principalmente a la aorta6,7.
De la misma forma, otros autores han elaborado revisiones sobre los diferentes patrones de alteraciones oculares basados en el fenotipo motor ocular; trabajos como el de Rucker en el que, de 40 sujetos, 37 tenían debilidad facial bilateral y 3 tenían debilidad facial unilateral. El patrón motor ocular más común (patrón 1, n=17, 43%) fue la parálisis mirada horizontal bilateral con rango vertical intacta. Patrón 2 (n=10, 26%) fue la parálisis mirada horizontal bilateral con limitaciones verticales variables. Patrón 3, que era raro: se aisló déficit de abducción (n=2, 5%). Otros tenían rango de movilidad completo y no cumplían con los criterios mínimos para el diagnóstico del MBS (modelo 4, n=10, 26%). Un sujeto presentaba ptosis bilateral severa, por lo cual fué imposible valorarlo con el patrón fenotipo motor ocular. Con el uso de esta clasificación de los patrones de déficits motores oculares y debilidad facial congénita, se puede ayudar con mayor delimitación a la localización anatómica y la identificación de los déficits genéticos que subyacen a estos trastornos8,9.
El problema de mayor relevancia en cuanto a la supervivencia de los niños con esta patología es que en los primeros meses la alimentación puede presentar dificultades enormes. En vez de ser una placentera y agradable satisfacción entre madre e hijo, la alimentación puede llegar a transformarse en una pesadilla. Porque para el bebé es difícil conseguir la cantidad de alimento requerido, además de frecuentes toses, vómitos y atragantamientos, que acaban produciendo miedo y rechazo hacia la comida en ambos, madre e hijo. La persistencia de un modo suave y amable es a menudo recompensada con el éxito. Sin embargo, algunas veces son necesarios meses para que el niño adquiera suficiente control y confianza para alimentarse placenteramente. Estas anomalías craneofaciales, maloclusiones y complicaciones dentales reconocidas en los pacientes que presentan este síndrome pueden ser resueltas según varios autores con el tratamiento de ortodoncia, en el que se utilizan aparatos removibles y cirugías reconstructivas; sin embargo, a pesar de una buena cooperación por parte del paciente, se observa solo una ligera mejora. Lo que se trata de evitar con este tratamiento es la extracción de los dientes permanentes y el uso de aparatos fijos mientras se espera la mejora de la oclusión y se evita la afectación de la articulación temporomaxilar10,11.
Aunque parece ser genético, su causa precisa aún es desconocida y las investigaciones médicas presentan teorías conflictivas. Algunos autores señalan que podría deberse a la ingesta de misoprostol, análogo semisintético de las prostaglandinas E1. Dado el uso de misoprostol para el aborto y la inducción de la actividad uterina en combinación con los antiinflamatorios no esteroideos, el número de recién nacidos con MBS asociados a este fármaco se ha incrementado. Hoy en día, tanto factores genéticos como ambientales se asocian con MBS; las estadísticas señalan que uno de cada 1.0000 nacimientos presenta este síndrome que afecta a niños y niñas por igual y, en algunos casos, parece haber un riesgo más alto de que se pase el trastorno de un padre afectado a su hijo o hija12.
Las bases moleculares del MBS son diversas; en un trabajo realizado por Kadakia et al. se proporciona un resumen exhaustivo de la literatura genética y etiológica subyacente a este trastorno. Para el efecto, seleccionaron y revisaron artículos desde 1880 hasta 2013, para entender todas las teorías moleculares y la cronicidad de los avances en la literatura, con la finalidad dilucidar las causas genéticas del trastorno y ayudar en la detección temprana y la planificación del tratamiento. Los resultados encontrados dieron que mutaciones en el gen loci MBS1, MBS2 y MBS3 han contribuido al desarrollo de MBS a través de diversas vías. Los genes HOX de la familia de codificación para los dominios homeobox, también, han sido implicados en el desarrollo anormal del cerebro humano. Así se obtuvieron hallazgos nacientes de los determinantes moleculares de MBS; los resultados de este estudio se suman a una base de datos cada vez mayor de mutaciones asociadas a MBS y se pueden utilizar para diagnosticar el MBS y aclarar la patogénesis. A pesar de que actualmente no existe una prueba prenatal para determinar el MBS, la gente se puede beneficiar del consejo genético13-15.
En el caso del paciente mencionado en este reporte, presenta parálisis facial, dificultad para la deglución en la lactancia y trastornos de los movimientos oculares y el cierre palpebral, más hipoplasia maxilar; estos signos y síntomas semejan a los presentados en los demás pacientes descritos en la literatura médica, ya que la mayoría de ellos son llevados a la consulta por un cuadro clínico compatible con el compromiso de los pares craneales vii, iii, xi y xii, observados en la valoración del recién nacido inmediatamente después del nacimiento.
Este caso presenta una variación de presentación; en la ecografía abdominopélvica y en la resonancia magnética se observó agenesia renal izquierda, con la presencia del riñón derecho ejerciendo un papel compensador.
Recomendaciones y conclusionesEn conclusión, se debe pensar en este diagnóstico, MBS, porque es frecuente y con formas de presentación muy amplias, pudiendo presentarse como una agenesia del pectoral mayor hasta un compromiso neurosensorial severo que lleve a la necesidad de un equipo multidisciplinario para su manejo. Existen muy pocos informes del síndrome y aún se discute el consejo genético en estos casos, dada la heterogeneidad del origen del cuadro.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciaciónNo contamos con financiamiento.
Conflicto de interesesLos autores no presentan conflictos de interés.
