



Sturge-Weber syndrome (SWS) or encephalotrigeminal angiomatosis is a sporadically presenting neuroectodermatosis characterised by a facial port-wine stain in the trigeminal distribution, ipsilateral leptomeningeal angioma, and ipsilateral choroidal angioma. Studies have described clinical variants with different combinations within this triad. SWS is primarily diagnosed in children, but it has also been diagnosed in adults in exceptional cases. We present a case of isolated leptomeningeal angiomatosis and symptom onset in an adult patient.
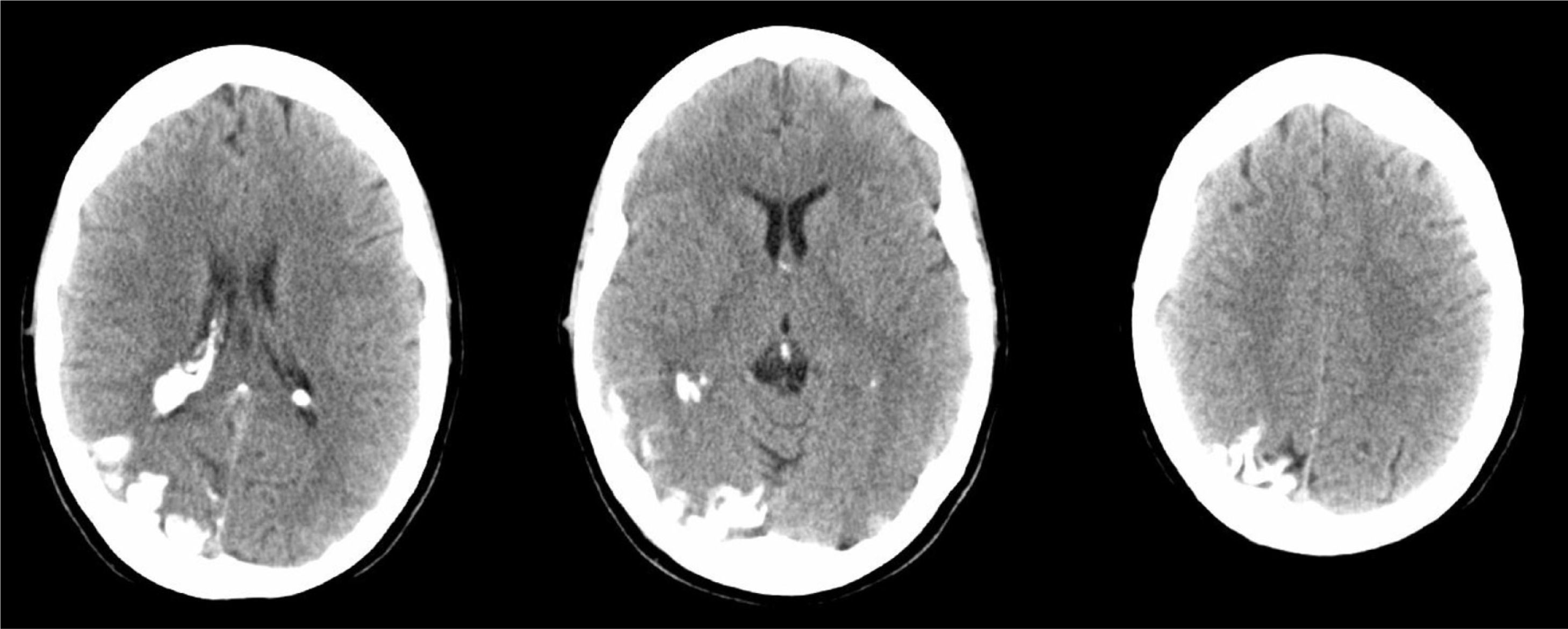
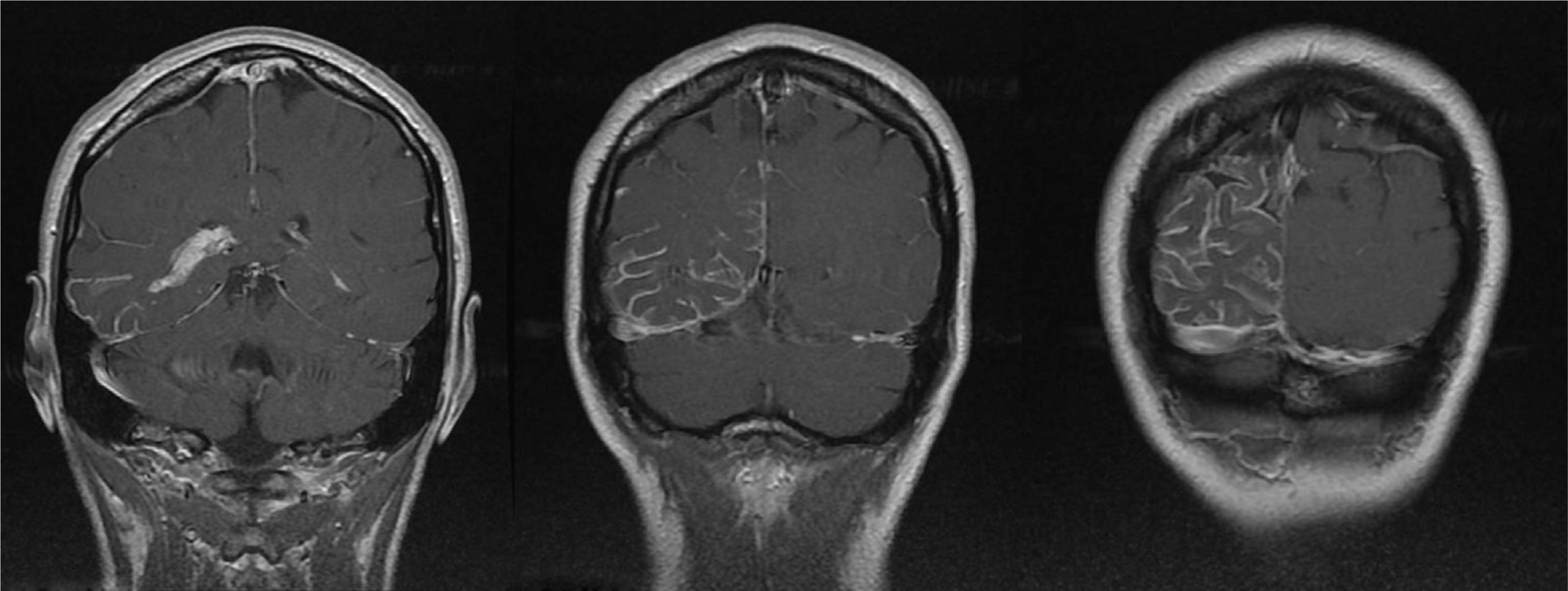
A 44-year-old woman with no relevant medical history came to the emergency department due to sudden-onset right hemicranial headache that was intense, oppressive, and responding poorly to analgesics. Twenty-four hours later, symptoms also included disorientation, psychomotor agitation, and visual hallucinations. Physical examination revealed a herpes labialis sore on her upper lip. Laboratory analyses revealed leukocytosis with left shift and elevated C-reactive protein (CRP). Cranial axial computed tomography with and without contrast (Fig. 1) showed a right parietal-occipital gyriform hyperdensity and focal meningeal enhancement that was initially reported as being compatible with chronic meningitis. Lumbar puncture yielded acellular CSF with elevated protein levels, so the emergency department started provisional treatment with IV vancomycin, ceftriaxone, and acyclovir for possible meningoencephalitis. The patient was admitted to the neurology department and remained afebrile with an intense headache accompanied by nausea, vomiting, and fluctuating episodes of visual and auditory hallucinations. Neurological examination detected homonymous hemianopsia. Brain MRI scans performed with and without gadolinium contrast (Fig. 2) showed leptomeningeal angiomatosis in the right hemisphere (parietal-occipital-temporal area). Basal amplitude on the electroencephalogram was attenuated in the right temporal-occipital area. There were no data indicating infection (results from 2 blood and CSF cultures and CRP tests were all negative); since a second lumbar puncture yielded acellular CSF with elevated opening pressure and protein levels, antibiotics were discontinued. A consultation with the ophthalmology department ruled out ocular vascular malformation. The patient was diagnosed with SWS without facial angioma and exhibiting temporal-occipital partial seizures and possible venous thrombosis/stasis at the angiomatous lesion. She was treated with acetazolamide (for intracranial hypertension), levetiracetam, and acetylsalicylic acid. Both the headache and the confusional episodes/hallucinations resolved; the latter were thought to be caused by seizure or vascular migraine-like phenomena. The patient subsequently attended follow-up visits as a neurology outpatient. After an additional lumbar puncture yielded acellular CSF with normal opening pressure and protein levels, the dose of acetazolamide was gradually reduced until it could be discontinued. The patient remained asymptomatic at a year of follow-up.

, but especially leptomeningeal. Size difference between choroid plexi of the temporal horns of the lateral ventricles; choroid plexi are larger and show more enhancement in the right lateral ventricle.")
Cranial MRI: axial T1-weighted contrast-enhanced images with striking meningeal enhancement mainly observed in the parietal-occipital-temporal regions. Enhancement in cerebral sulci was both dural and leptomeningeal (pial), but especially leptomeningeal. Size difference between choroid plexi of the temporal horns of the lateral ventricles; choroid plexi are larger and show more enhancement in the right lateral ventricle.
Encephalotrigeminal angiomatosis or SWS is an uncommon neurocutaneous syndrome affecting brain microvasculature that presents sporadically and is usually diagnosed in childhood. Clinical characteristics of its typical form include a flat facial angioma (port-wine stain) in the distribution of the ophthalmic branch of the trigeminal nerve, ipsilateral leptomeningeal angiomatosis at the level of the occipital and parietal lobes, and ocular vascular malformation. Incomplete variants of the syndrome1 include the following: (a) facial and leptomeningeal angioma, without choroid angioma; (b) leptomeningeal and choroid angioma without facial nevus; (c) facial nevus and choroid angioma with no clinical or imaging signs of cerebral angiomatosis; and d) isolated cerebral and pial angiomatosis.2–4
Although descriptions of SWS date back to 1879 (Sturge), Van Bogaert described a form of corticomeningeal telangiectasia without facial angioma as a variant of SWS in 1935. In Lund's 1949 review of the 144 reported patients affected by the syndrome, he identifies 7 cases with typical radiography images and no facial angiomas.5 The medical literature describes few cases of SWS without facial angioma,6–9 and a diagnosis in an adult patient, as in our case, is very rare.10
Neurological manifestations of SWS are related to the presence of leptomeningeal angioma.11 The most consistent sign is the presence of epileptic seizures, which affect 75% to 90% of all patients2; only 7% experience the initial seizure after the age of 5 years. Neurological deficits may appear slowly over time, or as stroke-like episodes associated with seizures and/or headaches resembling migraines as in the clinical case described here. The most frequently observed signs of neurological deficit are motor hemiparesis, hemianopsia, and hemiatrophy, which present in 65% of all patients. Cognitive changes are frequent and they are related to early onset of refractory seizures and bihemispheric dysfunction.12 Depressive symptoms are also common. Migraines and other headaches appear frequently, and this type is included in the second edition of the International Classification of the Headache Society (ICHD-II) among the secondary headaches in section 6.3.13
If there is a clinical suspicion of cerebral impairment, the condition is diagnosed using neuroimaging techniques, which provide very useful information in the absence of typical cutaneous or ocular signs. The characteristic radiography findings are gyriform cortical calcifications appearing adjacent to the leptomeningeal angioma in head CT images. Cranial MRI displays the typical vascular malformation; IV contrast provokes enhancement in T1-weighted images. In addition to these findings, we may also observe dilation and enhancement in the ipsilateral choroid plexus and enlargement of deep venous drainage underlying the affected cortical region.
Long-term treatment with antiepileptic drugs is fundamental for those patients with seizures. Since venous stasis and microvascular thrombosis probably contribute to the neurological decline occurring in SWS, the empirical recommendation calls for daily antiplatelet treatment with low doses of acetylsalicylic acid,14 which has been shown to decrease the frequency of seizures and stroke-like events. Headaches and migraines are treated with conventional abortive drugs (including triptans15); preventive drugs may also be used if required due to episode frequency or severity. Surgery (resection of the malformation or even hemispherectomy) provides effective treatment, but it should not be considered at time of diagnosis because it is currently indicated only in cases of seizures refractory to drug treatment.
The authors thank Dr. Noelia Arévalo Galeano at the Central Radiodiagnostics Unit for her assistance with this case.
Please cite this article as: Gómez-Moreno M, Murrieta-Urruticoechea C, Martinez-Acebes E, Gordo-Mañas R. Angiomatosis leptomeníngea temporo-occipital de diagnóstico en edad adulta. Neurología. 2015;30:64–66.
