



Las ataxias episódicas (AE), que se caracterizan por episodios recurrentes de incoordinación y desequilibrio, suponen una incidencia inferior a 1/100.0001. A pesar de presentar un cuadro clínico característico, su diagnóstico a veces puede retrasarse por la presencia de clínica interictal y por su asociación en un mismo paciente con otros trastornos neurológicos episódicos como la epilepsia, la discinesia paroxística y la migraña2.
Se presenta el caso clínico de un varón de 46 años, con hipertensión arterial y dislipemia, que refería episodios de horas de duración de disartria e inestabilidad. No refería antecedentes familiares de interés, sin posibilidad de consanguinidad familiar y siendo el mayor de 4 hermanos (3 hermanas de 30, 31 y 35 años) y padre de un hijo de 4 años. En la infancia había recibido tratamiento antiepiléptico por episodios de pérdida de consciencia. Asociaba cefalea episódica de características migrañosas. Veinte años antes había ingresado en otro centro durante uno de los episodios, realizándose resonancia magnética (RM) cerebral y cervical, electromiograma (EMG) y análisis sanguíneo incluyendo autoinmunidad y serologías, sin hallazgos, por lo que fue dado de alta encontrándose asintomático, sin alcanzarse un diagnóstico. El paciente describía que, si bien los episodios tenían una frecuencia variable, desde varios episodios el mismo día a ningún episodio en varias semanas, su frecuencia era mayor en situaciones de estrés o cansancio. El paciente aquejaba empeoramiento progresivo de la marcha interictal.
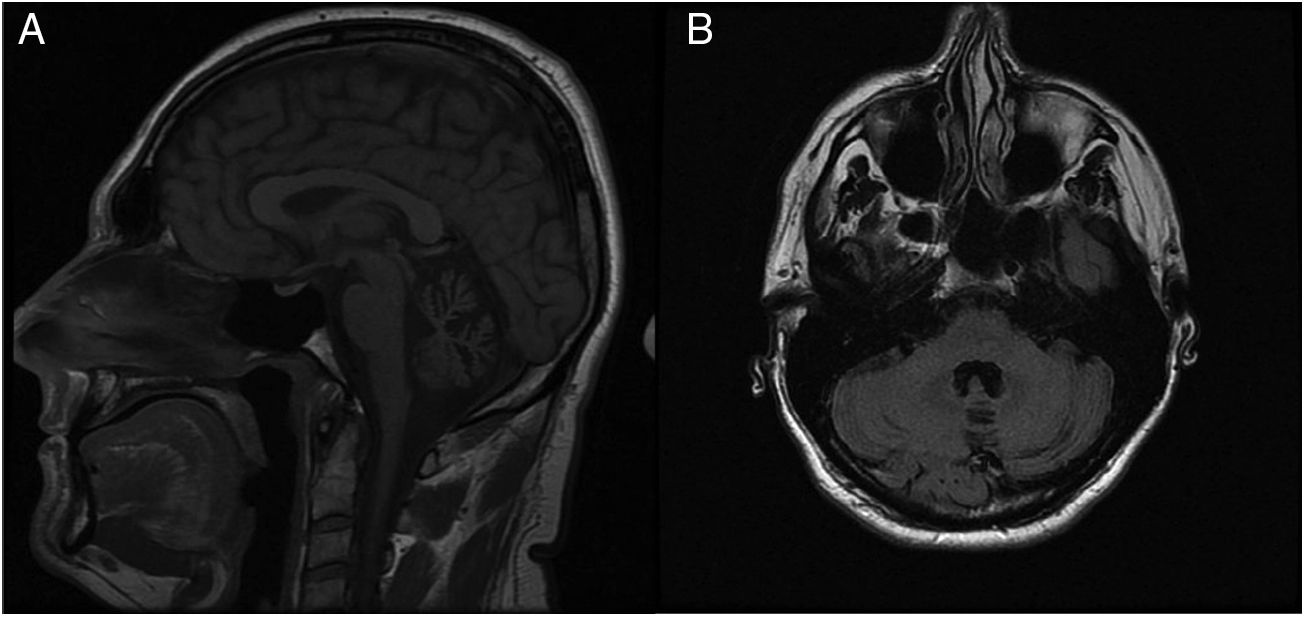
En la exploración neurológica interictal se objetivaron nistagmo horizontal multidireccional no agotable, disartria leve y dismetría dedo-nariz bilateral. Se había solicitado previamente estudio genético de ataxia espinocerebelosa, con resultados negativos para genes ATXN2 (SCA2) y ATXN3 (SCA3). Se revisó RM cerebral, informada como normal, en la que se objetivó atrofia de vermis cerebeloso (fig. 1).
 Corte sagital en secuencias T1. B) Corte axial en secuencias FLAIR.")
Dada la cronología de los episodios (autolimitados y de horas de duración) y la presencia de nistagmo y ataxia progresiva interictales, se solicitó estudio del gen CACNA1A, responsable de la AE tipo 2, en el que se detectó una variante missense c.889G>A (p.Gly297Arg;Het). Se prescribió acetazolamida, con reducción en la frecuencia de los episodios.
La AE tipo 2 es la más frecuente de los 8 subtipos de AE descritos hasta la fecha. Su primera descripción clínica data de 19463, pero no se descubrió su relación con el gen CACNA1A, que codifica para el canal de calcio P/Q dependiente de voltaje, hasta 19964. Tiene una herencia autosómica dominante5, con una penetrancia alta, pero incompleta6. Se conoce que la misma mutación del gen CACN1A puede presentar variabilidad fenotípica desde migraña con auras múltiples hasta déficits neurológicos focales transitorios sin cefalea, coma inducido por traumatismo craneoencefálico leve y ataxia cerebelosa lentamente progresiva7. En concreto, la mutación encontrada en nuestro paciente solo se ha descrito en una familia con episodios recurrentes de ataxia episódica y disfunción oculomotora y cerebelosa leve8, y en un niño con desviación tónica de la mirada hacia arriba que posteriormente desarrolló ataxia episódica, retraso del desarrollo psicomotor y migraña9.
A pesar de que inicialmente se trata de un trastorno episódico, con el estrés físico y emocional como desencadenantes más típicos, hasta el 90% de los pacientes desarrollan con el tiempo nistagmo interictal y el 50%, ataxia progresiva10. Se ha descrito que los pacientes con AE tipo 2, al igual que nuestro paciente, presentan un riesgo aumentado de epilepsia, y aproximadamente el 50%, migrañas episódicas. También se han documentado alteraciones en el EMG de fibra única con mejoría tras estimulación repetitiva, compatibles con afección de la transmisión neuromuscular a nivel presináptico11.
En la AE tipo 2 es frecuente encontrar atrofia cerebelosa vermiana en la RM cerebral, sobre todo en los casos con enfermedades de larga evolución con ataxia interictal persistente que, sin embargo, en ausencia de una sospecha clínica importante, puede pasar desapercibida en un primer momento como en nuestro caso.
El diagnóstico diferencial de las AE incluye causas periféricas de vértigo (vértigo posicional paroxístico, enfermedad de Ménière, enfermedad autoinmune del oído interno, laberintitis, etc.) y causas centrales como migraña, tóxicos, epilepsia, esclerosis múltiple, malformación de Chiari tipo 1, discinesias paroxísticas, trastornos funcionales y anomalías atloaxoideas6. La mayor parte de estas entidades pueden descartarse mediante una adecuada anamnesis y un estudio complementario. Sin embargo, la dificultad diagnóstica principal de la AE tipo 2 es con otras entidades relacionadas con el gen CACN1A, como la migraña hemipléjica familiar tipo 1 en la que es frecuente encontrar una disfunción cerebelosa progresiva indistinguible de la AE tipo 2 y la SCA6 con curso fluctuante y respuesta a acetazolamida10.
Creemos que nuestro caso clínico enfatiza la necesidad de considerar la AE tipo 2 en pacientes con episodios recurrentes de ataxia de horas de duración. Sorprende el retraso diagnóstico de más de 20 años en un paciente con datos clínicos típicos, lo que pensamos que pudo estar motivado por la presencia de ataxia y nistagmo interictales, la ausencia de antecedentes familiares y la historia clínica previa de epilepsia en la infancia.
FinanciaciónNo existe financiación para la realización de este trabajo.

