



La porfiria aguda intermitente (PAI) es una de las 4 porfirias agudas que se presenta con síntomas neurológicos o psiquiátricos. En adultos jóvenes sin antecedentes específicos puede comenzar en forma de una encefalopatía con alteraciones en la neuroimagen que pueden conducir a interpretaciones diversas. Los hallazgos típicos en la RM son los del síndrome de encefalopatía posterior reversible (SEPR) que se resuelve tras el tratamiento médico. Describimos el caso de una paciente joven con una PAI que se presentó como un inicio de epilepsia con alteraciones en la RM compatibles con SEPR.
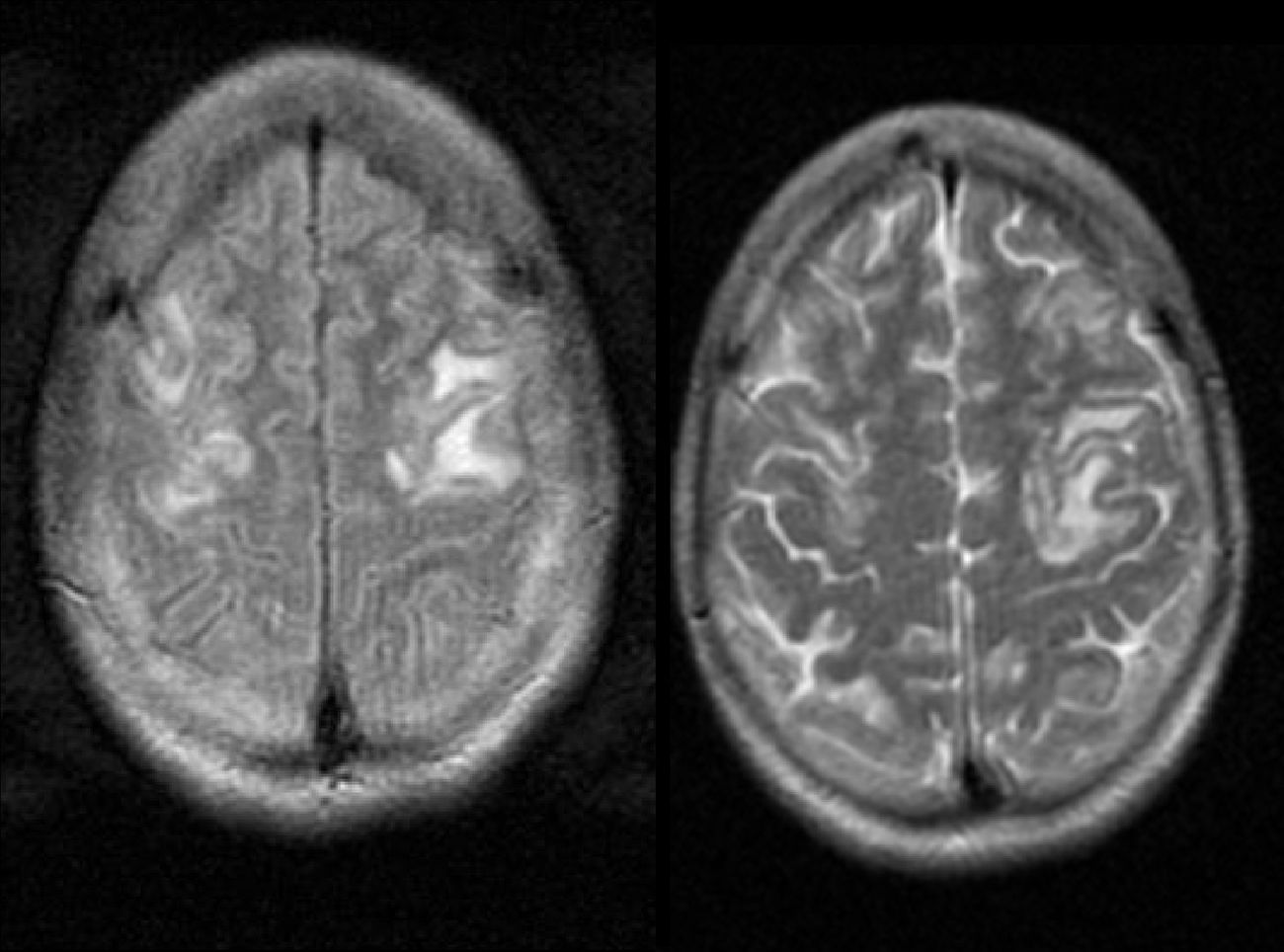
Se trata de una mujer de 23 años sin antecedentes de interés que presenta pérdida de conciencia seguida de 2 crisis tónico-clónicas que ceden tras tratamiento con midazolam. Al llegar a urgencias se realiza una tomografía computarizada craneal y analíticas, que eran normales, salvo una leve hiponatremia. Remitida a estudio ambulatorio con levetiracetam tras 24h de observación, vuelve al hospital al día siguiente tras sufrir otra nueva crisis y síntomas de desorientación y agitación que precisaron tratamiento con haloperidol. Se decide ingresar en Neurología para completar el estudio. Los estudios básicos bioquímicos y hematológicos no revelaron ninguna anormalidad (vitaminaB12, ácido fólico, coagulación y metabolismo del hierro), así como las serologías, hemocultivos, autoinmunidad y punción lumbar. Sin embargo, la paciente presentaba nuevos episodios de desorientación, agitación y fluctuación del nivel de conciencia, en una ocasión coincidiendo con una hiponatremia severa de 114mEq/l. Se realiza una RM que muestra lesiones frontales bilaterales corticales y subcorticales, que se mostraron hipointensas en las secuencias de T1 e hiperintensas en T2 (fig. 1) e hiperintensas en las imágenes de coeficiente de difusión aparente.

El diagnóstico diferencial que se planteó dado la clínica, la hiponatremia y la neuroimagen era de encefalitis viral/autoinmune o PAI. El diagnostico de PAI se confirmó con análisis de orina, donde se observó un aumento de los niveles totales de porfirina de 257μg/24h (normal 25-220), uroporfirina de 200μg/dl (normal <2μg/dl), porfobilinógeno (PBG) de 216mg/24h (normal <2,5mg) y ácido delta aminolevulínico (ALA) 105,2mg/24h (normal 0-3,4mg/24h). Se estudió mediante secuenciación directa el gen HMBS, detectándose en heterocigosis 2 variantes c.83 G>T (p.S28I) c.825+7 G>T (IVS12+7G>T).
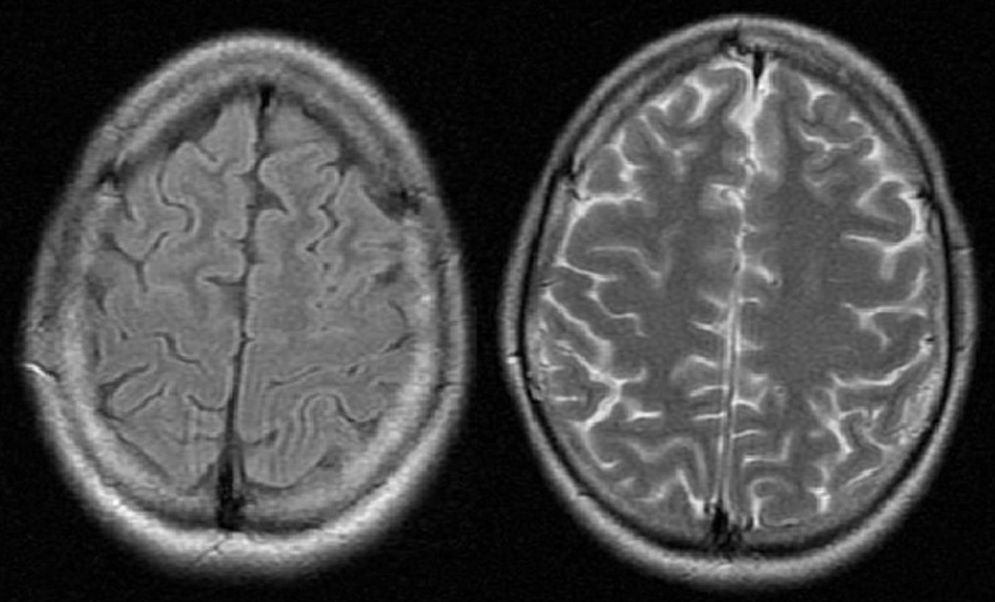
Se trató a la paciente con betabloqueantes y una dieta alta en hidratos de carbono, donde la paciente recuperó el nivel de consciencia con resolución de la desorientación y agitación sin secuelas. Se le siguió a los 2 meses del alta en las consultas de Neurología, estando estable con buen manejo de su nueva dieta, demostrándose normalización de las lesiones en RM (fig. 2).

Las porfirias son un grupo de enfermedades raras hereditarias donde hay una interrupción de la síntesis normal del grupo hemo de la hemoglobina. La PAI es la más común de las porfirias agudas, que incluyen también coproporfiria hereditaria (PH), porfiria variegata (PV) y porfiria deficiente de ácido delta aminolevulínico (ALAD-P)1. Pacientes con cualquiera de las porfirias agudas pueden comenzar con ataques severos que pueden ser precipitados por infección, alcohol, drogas, estrés, entre otros. La herencia de la PAI afecta tanto a hombres como a mujeres; sin embargo, su expresión es 5 veces mayor en el sexo femenino, sobre todo en la tercera década de la vida1,2. Las manifestaciones neurológicas son comunes, y en un 1% pueden llegar a ser mortales. La disfunción autonómica se manifiesta como dolor abdominal, náuseas, vómitos, diarreas, hipertensión y taquicardia. La afectación del sistema nervioso periférico puede dar parestesias, debilidad generalizada y parálisis respiratoria. Agitación, desorientación, ansiedad y convulsiones son producto de afectación del sistema nervioso central3,4. Las alteraciones de electrólitos son frecuentes en la PAI, siendo la hiponatremia la más común, ocurriendo en un 40% de los casos; esto se cree que se debe a una pérdida de sodio por el sistema gastrointestinal, sobrecarga de fluidos y por disminución de secreción de hormona antidiurética5.
El diagnóstico de la porfiria aguda es difícil, dados los síntomas variables y poco específicos. Si se sospecha, el análisis de orina es de suma importancia durante un ataque. Hay varias teorías del mecanismo de neurotoxicidad, como la acumulación de los precursores de heme en la neurona causando disfunción de la bomba Na+/K+ alterando el potencial de membrana causando degeneración y muerte axonal6.
En el diagnostico urinario de una porfiria aguda los niveles de ALA y PGB son eliminados más de 10 veces de los limites normales7,8, como el caso de nuestra paciente. En la PH y la PV estos niveles están simultáneamente incrementados, pero no tan elevados ni prolongados como en la PAI. Los niveles de ALA también se pueden ver en otras enfermedades metabólicas, como en el envenenamiento de plomo y en el ALAD-P8. En este último los niveles totales de porfirinas en orina no suelen estar elevados, diferenciando esta porfiria aguda de las otras tres. En la PH las coproporfirinas están muy elevadas, descartando este diagnóstico en nuestra paciente ya que eran normales. En la PV y la PH los síntomas suelen ser neuropsiquiátricos y cutáneos, mientras que nuestra paciente comenzó con crisis comiciales, dolor abdominal crónico, parestesias, hipertensión, sin lesiones cutáneas. Estos hallazgos clínicos y con los resultados urinarios apoyan más el diagnóstico de PAI.
El gold standard para el diagnóstico de las porfirias aguda es el test genético. En nuestra paciente la mutación era de significado incierto, ya que se detectaron 2 variantes que no se habían detectado previamente, la primera (c.83 G>T) produciendo un cambio significativo en la proteína y pudiendo ser causante de la enfermedad, mientras que la segunda (c.825+7 G>T) parece una variante polimorfa, lo que demuestra la heterogenicidad de las mutaciones que causan PAI.
La neuroimagen también es una herramienta de gran utilidad para el diagnóstico diferencial de PAI. La manifestación más frecuente es el síndrome de encefalopatía posterior reversible (SEPR)5, siendo una entidad tanto clínica y radiológica que se observan en el sistema nervioso central que tienen características típicas en la TC y en la RM2,4,7. Los hallazgos típicos en la neuroimagen son de edema vasogénico en la corteza; sin embargo, a pesar de su nombre las lesiones raramente se localizan exclusivamente en la parte «posterior» de la sustancia blanca de los lóbulos parietooccipitales, siendo más común la afectación de lóbulos frontales, ganglios basales y cerebelo8. En un estudio, la mayoría (54%) de los pacientes con SEPR que se detectaron en FLAIR y DWI tuvieron afectación frontal exclusivamente8, mientras que en otro estudio el 77% de los pacientes que tuvieron afectación frontal la mayoría también tenían afectación posterooccipital9. En la RM, las lesiones aparecen como iso- o hipodensas en secuencias de T1, e hiperintensas en T210,11. En el caso de nuestra paciente se observaron señales hipodensas en T1 e hiperintensas en T2 en ambos lóbulos frontales (figs. 1 y 2). Otra herramienta de gran utilidad es la RM de difusión, que ayuda a diferenciar el edema secundario a un proceso citotóxico, como en el infarto cerebral, del edema vasogénico12,13. En el caso de edema citotóxico las imágenes de RM (secuencia de difusión) muestran un aumento de la señal, mientras que en el edema vasogénico, como es el caso de SEPR, las imágenes se ven como iso- o hipodensas en RM14,15. Cabe resaltar que en el contexto de convulsiones intensas o prolongadas se han descrito cambios en la RM donde se observa hiperintensidad en áreas cortico-subcorticales en imágenes T2, indicando aumento de líquidos. Estas lesiones muestran restricción en la secuencia de difusión, apoyando el edema citotóxico más que vasogénico14.
La fisiopatología de SEPR aún no es del todo conocida. La hipótesis vascular indica que durante un ataque de porfiria hay vasoespasmo periférico, seguido de un aumento de la presión arterial, dando lugar a las manifestaciones cerebrales que semejan a las de la hipertensión maligna15,16. Otros mecanismos podrían ser los efectos neurotóxicos de un exceso de las porfirinas y sus precursores, o por un déficit de cofactores en la síntesis del grupo hemo16.
El diagnóstico diferencial de SEPR incluye isquemia cerebral aguda, trombosis venosa cerebral, encefalopatías pontinas en contexto de encefalopatías mitocondriales (MELAS), enfermedad de Creutzfeldt-Jakob y gliomatosis cerebral17,18. Estas entidades se pueden diferenciar con la historia clínica, la exploración, los hallazgos de laboratorio y la neuroimagen.
Nuestro caso clínico ayuda a concienciarnos de que la PAI puede comenzar de una manera insidiosa con afectación neurológica, siendo esta común y muy variable. La RM es una herramienta útil para diagnosticar las porfirias agudas que típicamente se manifiesta con el SEPR que remite con el tratamiento adecuado y con mejoría de la sintomatología.
FinanciaciónLos autores declaramos que no hubo ningún tipo de financiación pública o privada para la realización del presente reporte de caso clínico.
Conflicto de interesesDeclaramos que no existe ningún tipo de conflicto de intereses en el presente manuscrito.

