



Cerebrovascular diseases are among the leading causes of death and disability in developed countries. Acetylsalicylic acid (ASA) and clopidogrel are the most widely used antiplatelet drugs for secondary prevention of recurrent thromboembolic events. However, there have been cases in which antiplatelet drugs did not inhibit platelet activity; this phenomenon is called resistance, and it may be modulated at the genetic level.
DevelopmentFollowing a literature search, we reviewed the current state of antiplatelet therapy and covered the different types of resistance to antiplatelet therapy, how it is measured, current problems and limitations, and any genetic factors that have been associated with resistance. We mainly used the Genome Wide Association Studies in the field of ASA and clopidogrel resistance.
ConclusionsWe observed an association between different genetic factors and antiplatelet drug resistance as measured by platelet activity. However, there is no evident association between these genetic factors and risk of new thromboembolic events.
Las enfermedades cerebrovasculares están entre las principales causas de mortalidad y discapacidad en los países desarrollados. El ácido acetilsalicílico (AAS) y el clopidogrel son los tratamientos antiagregantes plaquetarios más utilizados para la profilaxis de nuevos eventos tromboembólicos. Sin embargo, se han observado casos en los que el tratamiento antiagregante no inhibe la actividad plaquetaria, un fenómeno llamado resistencia y que posiblemente puede estar modulado a nivel genético.
DesarrolloTras una búsqueda bibliográfica se realizó una revisión sobre el estado actual del tratamiento antiagregante plaquetario. Se tratan los diferentes tipos de resistencia a la terapia antiagregante, de qué manera se mide, la problemática y limitaciones actuales, así como los factores genéticos que se han asociado a esta resistencia. Principalmente se analizan los estudios genéticos realizados en el campo de la resistencia a AAS y clopidogrel mediante Genome Wide Association.
ConclusionesParece existir una asociación entre diferentes factores genéticos y la resistencia a los fármacos antiagregantes medida mediante la actividad plaquetaria; no obstante, no hay una asociación evidente entre estos factores genéticos y el riesgo de nuevos eventos tromboembólicos.
Stroke is the second most common cause of mortality worldwide and the leading cause of disability in developed countries. It accounted for 6.2 million deaths in 2008 and its incidence rate was 200 new cases per 100000 inhabitants per year.1
After the first episode, the cumulative risk of recurrence is 11.1% in the first year, 26.4% during the following 5 years, and 39.2% at 10 years.2 Viewed by subtype, atherothrombotic and cardioembolic strokes present a higher risk of recurrence than lacunar strokes.3 Platelets are largely responsible for thrombus formation. Platelet activation is mediated by a number of factors, including serotonin, epinephrine, adenosine diphosphate (ADP), and thromboxane A2. Antithrombotic or antiplatelet drugs are some of the most widely used types of drugs for secondary prevention of ischaemic stroke since they help halt new vascular episodes.4 Antiplatelet drugs, especially acetylsalicylic acid (ASA) and clopidogrel, are the most commonly prescribed drugs for secondary prevention of ischaemic stroke, except in patients with cardioembolic ischaemic stroke treated with anticoagulants and who have no risk of haemorrhage.3
Despite the obvious benefits of antiplatelet drugs, platelet reactivity varies greatly and patients do not respond to antiplatelet therapy in a uniform way. Risk reduction achieved with these drugs has been found to be suboptimal. For example, patients’ mortality rates show a decrease of 18% whereas disability rates decrease by 25%. In fact, 10% to 20% of patients treated with ASA or clopidogrel experience new vascular events.5 This may be due to the development of drug resistance. Resistance is defined as the presence of normal platelet activity despite correct administration of antiplatelet therapy. Different strategies and drug combinations have been used to lower vascular recurrence rates, without generating promising results.6–8
The present review describes the genetic studies conducted in the field of antiplatelet drug resistance and addresses the problems arising from associating resistance, measured by platelet activity, with the onset of new vascular events.
DevelopmentWe conducted a literature search on PubMed and Google Scholar using the keywords ‘stroke’, ‘aspirin’, ‘clopidogrel’, ‘antiplatelet therapy’, ‘resistance’, ‘genetics’, and ‘pharmacogenomics’. We only considered articles from indexed scientific journals, and selected original and review articles published in either English or Spanish.
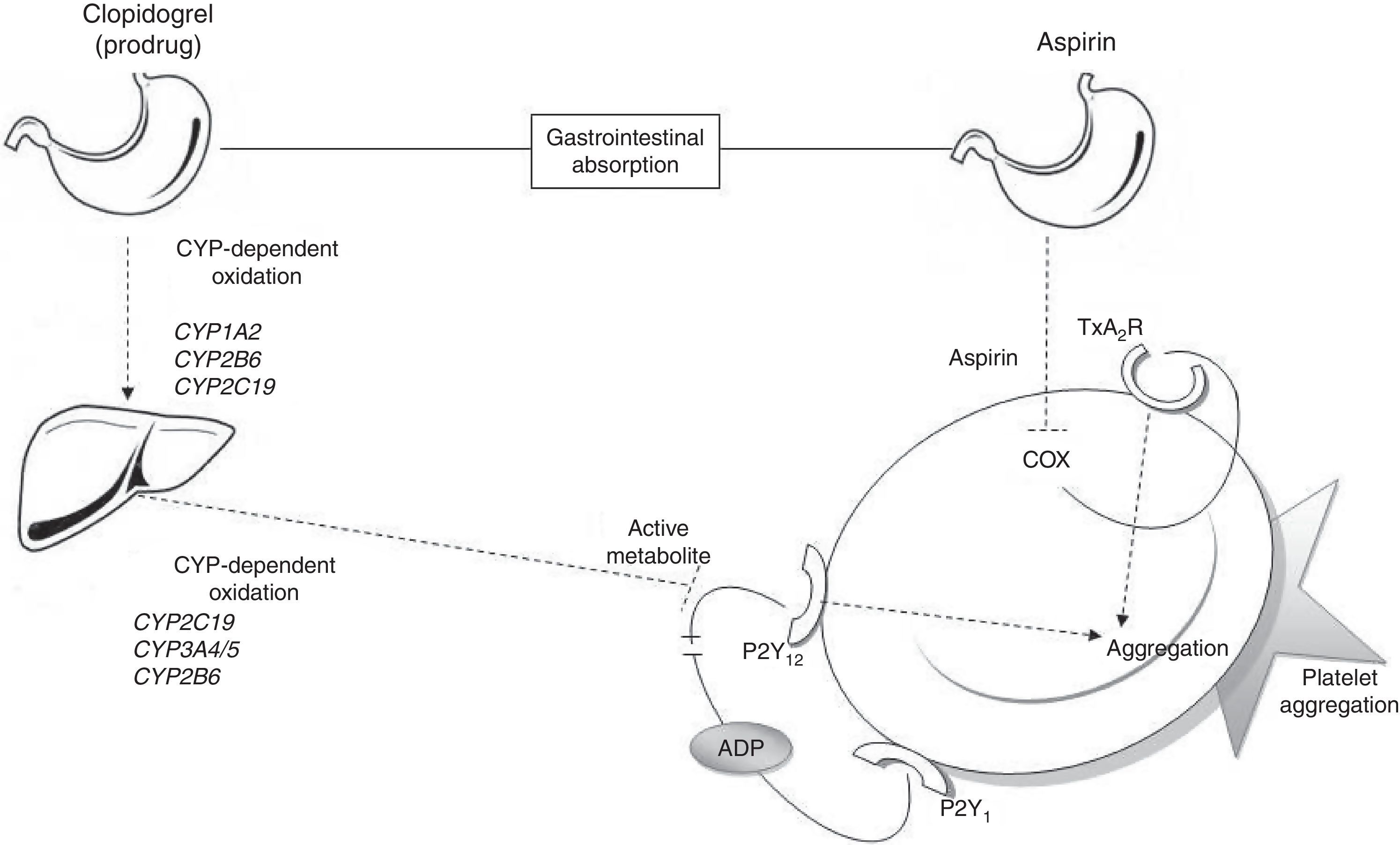
Metabolic pathways for acetylsalicylic acid and clopidogrelThe antiplatelet effect of ASA is achieved through permanent inhibition of the cyclooxygenase enzyme (COX).6 COX has 2 different isoforms: COX-1, a constitutive isoform present in most cells, and COX-2, which is expressed in response to inflammatory stimuli. ASA irreversibly inhibits COX-1 by blocking arachidonic acid conversion to thromboxane A2 (Fig. 1); this way it blocks the main triggering factor of platelet activation.6

Connection between antiplatelet drugs and platelet aggregation. Metabolism of clopidogrel: clopidogrel is metabolised in the liver in 2 oxidation stages which involve several CYP enzymes. The active metabolite inhibits binding of ADP to the P2Y12 receptor, thus blocking platelet aggregation. Metabolism of ASA: ASA is absorbed by the stomach and small intestine. It blocks platelet aggregation by inhibiting COX activity and thromboxane production. ADP: adenosine diphosphate; COX: cyclooxygenase; CYP: cytochrome P450; TxA2R: thromboxane A2 receptor.
Clopidogrel, however, is a prodrug that must be metabolised in the liver in order to be transformed into an active metabolite (thiolactone) with antiplatelet activity. This process has 2 oxidative steps and is mediated by different hepatic enzymes of cytochrome P450 (CYP), including CYP1A2, CYP2B6, CYP2C19, and CYP3A4/5. The antithrombotic effect of clopidogrel is due to the irreversible binding of thiolactone to the ADP receptor (P2Y12) on the platelet surface (Fig. 1).7
Resistance to acetylsalicylic acid and clopidogrelPatient responses to antiplatelet therapy vary greatly.7,9 Resistance can be detected either using biochemical analyses or clinically (new or recurring vascular events). Although these concepts relating to resistance are apparently similar, they do present differences. A patient shows resistance to antiplatelet drugs when these drugs fail to inhibit platelet aggregation, a situation that can be detected with functional biochemical analyses. When these functional tests register normal platelet activity, the condition is commonly known as drug resistance. On the other hand, clinical resistance (or treatment failure) is defined as onset of a cardiovascular event despite correct administration of and adherence to treatment. The association between drug resistance measured by a biochemical analysis and occurrence of new vascular events (or clinical resistance) remains unclear.
Functional tests for studying resistance to ASA and clopidogrel are based on platelet aggregation studies, especially kinetic studies. There are multiple techniques for evaluating platelet aggregation, including optical aggregometry, impedance aggregometry, thromboxane A2 production, the platelet function analyzer-100 system (PFA-100), Ultegra RPFA-ASA, thromboelastography platelet mapping systems, and studies of activation-dependent changes on the platelet surface.9 Unfortunately, the results obtained with these techniques correlate poorly between one another, both in patients treated with ASA and in those taking clopidogrel.9 To date, there is no consensus on the most suitable biochemical test for measuring resistance to antiplatelet therapy.
Around 60% of patients are treated with antiplatelet drugs after stroke.3,10 Knowing the factors that cause resistance is therefore essential for proper management of resistant patients, adjusting the dose, or changing the treatment if necessary. Various genetic studies aimed at elucidating the connection between patients’ genotypes and resistance to antiplatelet therapy have been launched in the past few years.
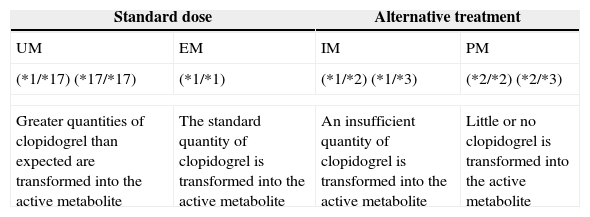
Classifying patients as rapid or slow metabolisersAs stated previously, clopidogrel needs to be transformed into an active metabolite by CYP enzymes in order to have an antiplatelet effect. The genes coding for CYP enzymes are polymorphic, with several alleles conferring an altered function.11 Depending on their CYP2C19 genotypes, individuals can be classified by phenotype as extensive metabolisers (EM), intermediate metabolisers (IM), or poor metabolisers (PM). Individuals carrying the CYP2C19*17 allele are categorised as ultrarapid metabolisers (UM). Ultrarapid metabolism of clopidogrel results in an increase in platelet inhibition and a subsequent reduction in residual platelet aggregation (the CYP2C19*17 allele responsible for this scenario may be associated with increased risk of bleeding). EMs are individuals with normal platelet inhibition and normal residual aggregation. IMs display reduced platelet inhibition and increased residual platelet aggregation. These individuals may also present an increased risk of adverse cardiovascular events. PMs typically show a significant reduction in platelet inhibition and, as a result, may be at greater risk for adverse cardiovascular events (Table 1).12
Clinical results by CYP2C19 genotype.
| Standard dose | Alternative treatment | ||
|---|---|---|---|
| UM | EM | IM | PM |
| (*1/*17) (*17/*17) | (*1/*1) | (*1/*2) (*1/*3) | (*2/*2) (*2/*3) |
| Greater quantities of clopidogrel than expected are transformed into the active metabolite | The standard quantity of clopidogrel is transformed into the active metabolite | An insufficient quantity of clopidogrel is transformed into the active metabolite | Little or no clopidogrel is transformed into the active metabolite |
CYP2C19 is a highly polymorphic gene. The most common functional polymorphisms are *2, *3, and *17; CYP2C19*1 is the normal variant. The result of enzyme activity depends on the genotype and is highly variable. This result has an impact on the individual's capacity to activate clopidogrel.
UM: ultrarapid metaboliser; EM: extensive metaboliser; IM: intermediate metaboliser; PM: poor metaboliser.
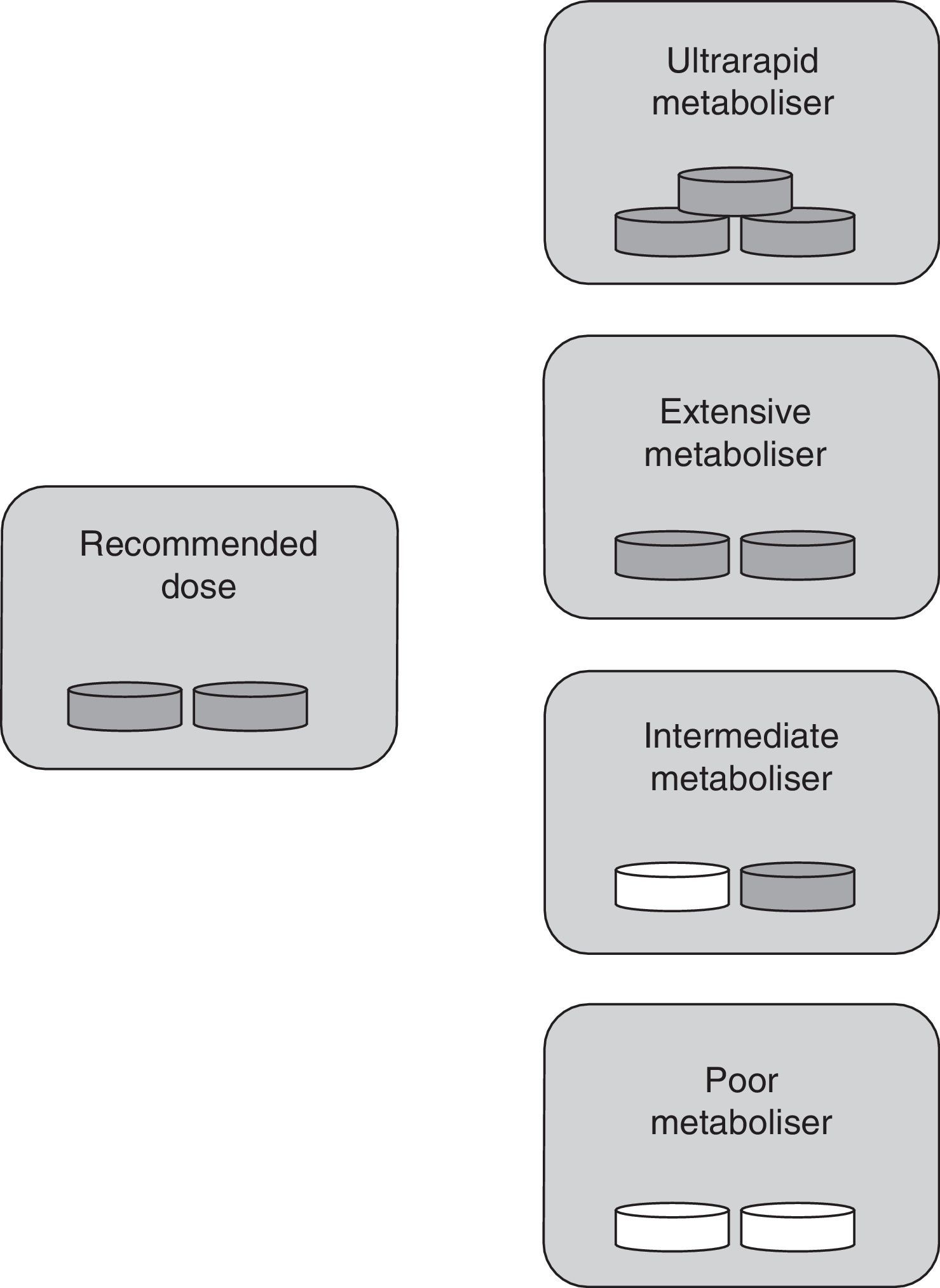
The classification into types of metabolisers for ASA differs from the one used for clopidogrel since ASA is already active when consumed. In this case, UM individuals with higher-than-normal enzymatic activity show ASA plasma levels that are lower than normal. This results in insufficient inhibition of platelet aggregation by the drug. These patients are classified as resistant to antiplatelet therapy. In IM and PM phenotypes, enzymatic activity is lower than expected; as a result, ASA accumulates in the blood, which may lead to toxicity. EMs therefore remain as the only individuals receiving antiplatelet therapy in its intended doses (Fig. 2).
, extensive metabolisers should be given standard doses, and intermediate and poor metabolisers require lower doses or changing to a different drug. Grey tablet: drug administered; white tablet: drug not administered.")
Specific drug dose by type of metaboliser for ASA. Ultrarapid metabolisers need more than the normal dose (they are treatment-resistant), extensive metabolisers should be given standard doses, and intermediate and poor metabolisers require lower doses or changing to a different drug. Grey tablet: drug administered; white tablet: drug not administered.
Pharmacogenetics is a field of genetics that analyses genetic variations that may affect responses to pharmacological treatments.
Candidate gene-based pharmacogenetic studies have found polymorphisms associated with resistance to ASA and clopidogrel. The candidate gene approach focuses on studying genes and polymorphisms which, because of their function, are thought to be associated with a specific disease. Polymorphisms are allele variants occurring in the same population. Each one is present in more than 1% of the population and it may be associated with distinct phenotypic traits, such as different responses to drugs. The candidate gene approach is biased since it analyses a reduced number of polymorphisms of a selected group of genes. Nevertheless, it has been shown to be useful for certain diseases and in some pharmacogenetic studies. Regarding ASA resistance, several studies have identified single nucleotide polymorphisms (SNP) in the genes COX-1, COX-2, GPIa, GPIbα, GPIIIa, GPIV, FXIII, P2Y1, and P2Y1213 as being associated with aspirin resistance. However, none of these studies included sufficiently large sample sizes or was replicated in an independent cohort to prove the association between these genes and aspirin resistance.
The first candidate gene-based studies of clopidogrel found polymorphisms associated with clopidogrel resistance in CYP genes, especially in the 2C19 subclass (CYP2CI9), but also in CYP2C9, CYP2B6, CYP3A4, CYP3A5, and CYP1A2. Other genes with SNPs linked to clopidogrel resistance are P2RY12, ABCB1, and PON1,11,14 although these results have not been validated by subsequent studies.
Genome-wide association studies of resistance to antiplatelet drugsGenome-wide association studies (GWAS) constitute one of the most widely used approaches in the field of genetics. GWAS are usually large-scale case-control studies which analyse a high number of polymorphisms (10000-2000000), generally SNPs, in order to determine which SNPs are associated with a specific phenotype. These studies offer an unbiased hypothesis-free approach to the study of complex diseases.15
Thanks to GWAS, genes have been linked to several complex diseases not previously known to have a genetic basis. For instance, these studies have found 2 loci (the genes PITX2 and ZFHX3) which are associated with cardioembolic ischaemic stroke. SNPs have also been found in genes CDK2MA and CDK2MB on chromosome 9 (9p21 locus),16 as well as in HDAC9 on chromosome 7 (7p21.1 locus),17,18 which are associated with atherothrombotic stroke.
Only one GWAS has been conducted to test resistance to clopidogrel; this study included 429 healthy Amish subjects who were treated with the drug for 7 days.19 Response to therapy was measured using ex vivo platelet aggregometry. The platelet response to clopidogrel was largely hereditary (h2=0.73; P<.001); 13 SNPs on chromosome 10 (10q42 locus), within the CYP2C18-CYP2C19-CYP2C9-CYP2C8 cluster, were found to be associated with a decreased response to clopidogrel. The rs12777823 polymorphism showed the most significant association with platelet activity (P=1.5×10−3). The rs12777823 SNP was in linkage disequilibrium with the CYP2C19*2 variant (r2=0.87), a polymorphism that had already been linked to clopidogrel resistance by candidate gene-based studies (Table 2).19 Shuldiner et al. replicated their GWAS in a new cohort. In this stage, they analysed the CYP2C19*2 variant in linkage disequilibrium with rs12777823 in 227 patients undergoing a coronary intervention. Patients carrying the risk-associated variant CYP2C19*2 presented more vascular events during the first year after the intervention.
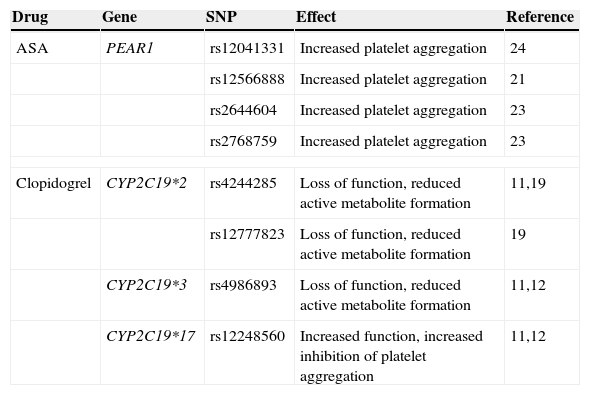
Main identified polymorphisms associated with platelet aggregation.
| Drug | Gene | SNP | Effect | Reference |
|---|---|---|---|---|
| ASA | PEAR1 | rs12041331 | Increased platelet aggregation | 24 |
| rs12566888 | Increased platelet aggregation | 21 | ||
| rs2644604 | Increased platelet aggregation | 23 | ||
| rs2768759 | Increased platelet aggregation | 23 | ||
| Clopidogrel | CYP2C19*2 | rs4244285 | Loss of function, reduced active metabolite formation | 11,19 |
| rs12777823 | Loss of function, reduced active metabolite formation | 19 | ||
| CYP2C19*3 | rs4986893 | Loss of function, reduced active metabolite formation | 11,12 | |
| CYP2C19*17 | rs12248560 | Increased function, increased inhibition of platelet aggregation | 11,12 | |
This and other studies recently led the US Food and Drug Administration to recommend genotyping to rule out the CYP2C19*17 polymorphism before prescribing clopidogrel (Table 2).20 This recommendation has generated a great deal of controversy since the American Heart Association and the American College of Cardiology state that data are insufficient to justify routine testing.21 The last update on this subject, published by the Clinical Pharmacogenetics Implementation Consortium (CPIC) in May 2013,22 suggests genotyping CYP2C19 before initiating antiplatelet therapy, especially in patients with acute coronary syndrome undergoing percutaneous coronary intervention. This recommendation is based on the finding that most recurrent and potentially foreseeable events take place at treatment onset. CYP2C19 genotyping may therefore be useful for adapting treatments to individual patients to avoid recurrence.
One GWAS, aiming to evaluate ASA resistance, was carried out in 2 cohorts: 2753 subjects from the Framingham Heart Study and 1238 subjects from the Genetic Study of Atherosclerosis Risk. Resistance to ASA was measured using biochemical tests. The study found 7 loci with polymorphisms associated with platelet aggregation. Results with a significance level of P<.05 were replicated in a cohort of 840 patients from the Genetic Study of Atherosclerosis Risk. One of the most interesting genes to be identified was PEAR1 (rs12566888, P=3.4×10−12), which was confirmed to be associated with resistance to ASA (Table 2).23
PEAR1, or platelet endothelial aggregation receptor-1, codes for a platelet transmembrane protein that is activated by contact with other platelets. An earlier study by Herrera-Galeano et al.,24 which genotyped 10 SNPs of PEAR1 in 1486 healthy individuals, found that the rs2768759 polymorphism was associated with increased platelet aggregation after treatment with ASA (Table 2).
A more recent GWAS was conducted by Lewis et al.25 to measure platelet aggregation before and after treatment with aspirin in 565 patients from the Pharmacogenomics of Anti-Platelet Intervention (PAPI) study. Results revealed a strong association between SNPs on chromosome 1q23.1 and response to ASA. The most significant findings were genotyped in 2 new cohorts, one comprising 227 patients undergoing coronary intervention and the other, 1000 patients from the genetic substudy within the International Verapamil SR/Trandolapril Study (INVEST-GENES). With these replications in independent cohorts, the SNP rs12041331 of PEAR1 was shown to have the strongest association with the response to antiplatelet therapy (P=7.66×10−9). These results were confirmed by the study by Kim et al.,26 who sequenced PEAR1 in 104 subjects with hypo- or hyperaggregation. The rs12041331 polymorphism was found to have the most significant association with the response to platelet aggregation (P=4.02×10−4).
Resistance measured with biochemical tests vs vascular recurrence or clinical resistanceDespite efforts to identify the genetic factors causing resistance to antiplatelet therapy, the evidence remains inconclusive. The association between vascular recurrence (clinical resistance) and platelet activity measured with the techniques described above is yet to be determined.27–29 As a consequence, the polymorphisms that have been linked to platelet activity are not clearly associated with vascular recurrence.27,29 In the genetic substudy in Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA), a trial which genotyped 4819 stable patients who had experienced a stroke or myocardial infarction and were treated with clopidogrel, polymorphisms rs11188078 and rs12248560 of the allele CYP2C19*17 were found to have no effect on the rate of ischaemic or haemorrhagic events.30
According to a more recent meta-analysis of 32 studies including a total of 42016 patients treated with clopidogrel and 3545 cardiovascular events, which is one of the most important pharmacogenetic studies on resistance to clopidogrel, CYP2C19 alleles were associated with vascular recurrence. However, the association between CYP2C19 polymorphisms and new vascular events was non-significant when the analysis only included studies with at least 200 subjects experiencing vascular recurrence. The meta-analysis therefore concluded that evidence was insufficient to support an association between CYP2C19 and vascular recurrence.30
There are several explanations for the lack of association between platelet activity (resistance) and vascular recurrence, on the one hand, and between the polymorphisms associated with platelet activity and vascular recurrence, on the other.
Additional problems are related to patient follow-up. One of the most common problems is patients’ lack of adherence to treatment, and secondarily, any changes applied to antiplatelet therapy during follow-up. Either of these cases may induce bias or reduce the statistical power of the study. Given the presence of these biases, SNPs associated with resistance to antiplatelet therapy may not be associated with new vascular events.
At the genetic level, there are several possible explanations for this lack of association between vascular recurrence and the polymorphisms associated with antiplatelet drug resistance. One reasonable explanation is that although platelet activity affects vascular recurrence, this association is weaker than expected. As a result, the polymorphisms associated with platelet activity are not significantly associated with vascular recurrence.
We need studies with larger population samples and better designs and follow-up procedures to answer the questions that remain, as well as studies including patients with clinical resistance or treatment failure, that is, patients experiencing vascular recurrence despite appropriate use of antiplatelet therapy.
On the other hand, additional genetic factors that have yet to be explored may be associated with resistance as measured by platelet activity and also with vascular recurrence. This may be true of rare variations, epigenetic factors, or alterations in the number of copies that have not been detected due to a small sample size or to lack of appropriate tools.
FutureThe studies conducted to date have not provided sufficient information to determine the genetic component of resistance to antiplatelet therapy. Further studies and new approaches are necessary in this field. The next few years are likely to bring novel epigenetic and sequencing studies examining the pharmacogenetics of antiplatelet drug resistance.31
New sequencing techniques are less expensive and provide better and faster results than traditional Sanger sequencing.30 These techniques are increasingly being used to confirm loci detected by GWAS and to identify genomic structural variations. Epigenetic studies will enable the study of gene expression mechanisms independent from the DNA sequence in patients resistant to antiplatelet drugs.32
Developing bioinformatics tools to analyse the massive amounts of data generated with these techniques will play a key role in achieving rapid advances in this field.32 Another crucial undertaking related to pharmacogenetic studies of complex diseases is the creation of consortia granting researchers access to greater sample sizes and helping establish more defined phenotypes. Not only will this step increase the statistical power of the studies, but it will also enable detection of new and currently undiscovered SNPs. The consortia that are addressing the area of antiplatelet drug resistance are as follows: the International Clopidogrel Pharmacogenomics Consortium, which aims to increase the number of subjects and conduct genetic studies on subjects with more rigorous clinical follow-up; CPIC (http://www.pharmgkb.org/page/cpic); the International Stroke Genetics Consortium (http://www.strokegenetics.org/); and the Spanish Stroke Genetics Consortium or GeneStroke (http://www.genestroke.com/). Since the new techniques generate a large quantity of data, online repositories and databases are gaining an increasingly important role. Some of the most important ones in this area are PharmGKB (http://www.pharmgkb.org/), which provides information on the impact of genetic variations on drug responses; dbSNP (http://www.ncbi.nlm.nih.gov/SNP/), a public archive of genetic variations across different species; and dbGaP (http://www.ncbi.nlm.nih.gov/gap), a database of phenotypes and genotypes. The Genetic Association Database (http://geneticassociationdb.nih.gov/), containing genetic association data, is especially interesting for other GWAS.
ConclusionsIncreasing our knowledge about drugs used in preventing new vascular events is very likely to have a major impact on health. Future genetic studies will need to make use of the new genomic techniques and be conducted within international consortia to better define patients’ phenotypes and increase sample sizes with the goal of completing large-scale projects that may help shed some light on this topic. Studies conducted to date have failed to show a definitive connection between vascular recurrence and the polymorphisms associated with clinical resistance. Prospective clinical trials will be necessary to prove this association, which would support use of genotyping as a standard tool in clinical practice. In the case of clopidogrel resistance, however, published studies seem to point to a connection between certain polymorphisms and platelet activity. Patients who are likely to experience drug resistance due to the presence of risk-associated CYP450 polymorphisms may be treated with a new antiplatelet drug, either individually or combined with a new antiplatelet drug (clopidogrel+ASA).
FundingThis study was funded by the Miguel Servet programme (CP12/03298) at the Institute of Health Carlos III. I. Fernández-Cadenas has been funded by the Miguel Servet programme (CP12/03298) at the Institute of Health Carlos III.
Conflicts of interestThe authors have no conflicts of interest to declare.
The Neurovascular Pharmacogenomics and Genetics Laboratory forms part of the International Stroke Genetics Consortium and the Spanish Stroke Genetics Consortium (www.genestroke.com).
Please cite this article as: Gallego-Fabrega C, Krupinski J, Fernandez-Cadenas I, en nombre de Genestroke Consortium, Consorcio Español para el Estudio Genético del Ictus. La resistencia en el tratamiento secundario del ictus isquémico, el componente genético en la respuesta a ácido acetilsalicílico y clopidogrel. Neurología. 2015;30:566–573.
