




La enfermedad de Creutzfeldt-Jakob (ECJ) es una enfermedad neurodegenerativa con curso rápidamente progresivo y pronóstico infausto. Se estima que solo el 10% de los casos son familiares (ECJf). En estos, existe una mutación en el gen de la proteína priónica, localizado en el cromosoma 20, donde puede haber codificada una metionina (M) o valina (V) en el codón 1291,2. No existen muchos casos documentados de ECJf en España. A continuación se presentan dos casos y la genealogía de una familia con la mutación E200K 129M/M.
Caso clínico 1Mujer de 59 años sin antecedentes relevantes que consulta por deterioro cognitivo, y trastorno de la marcha de un mes de evolución. La paciente desarrolla mutismo acinético y fallece 2 meses después del inicio del cuadro. Sus características clínicas se exponen en la tabla 1. Una tomografía por emisión de positrones con 18-fluorodesoxiglucosa (FDG-PET) a los 45 días del inicio del cuadro, muestra hipometabolismo en corteza parietal y estriado derecho (fig. 1A); y la resonancia magnética (RM) muestra hiperintensidad bilateral de predominio en zona insular y parietal derecha en secuencias Fluid-Attenuated Inversion Recovery (FLAIR) y Diffusion-Weighted Imaging (DWI) (fig. 1B). A los 60 días, un electroencefalograma (EEG) registra descargas focales punta onda con predominio para sagital derecho (fig.1C). La proteína 14-3-3 en el líquido cefalorraquídeo es positiva y el estudio genético confirma la mutación E200K 129M/M. En el estudio posmorten se observan cambios espongiformes en territorio subcortical y cortical de predominio izquierdo.
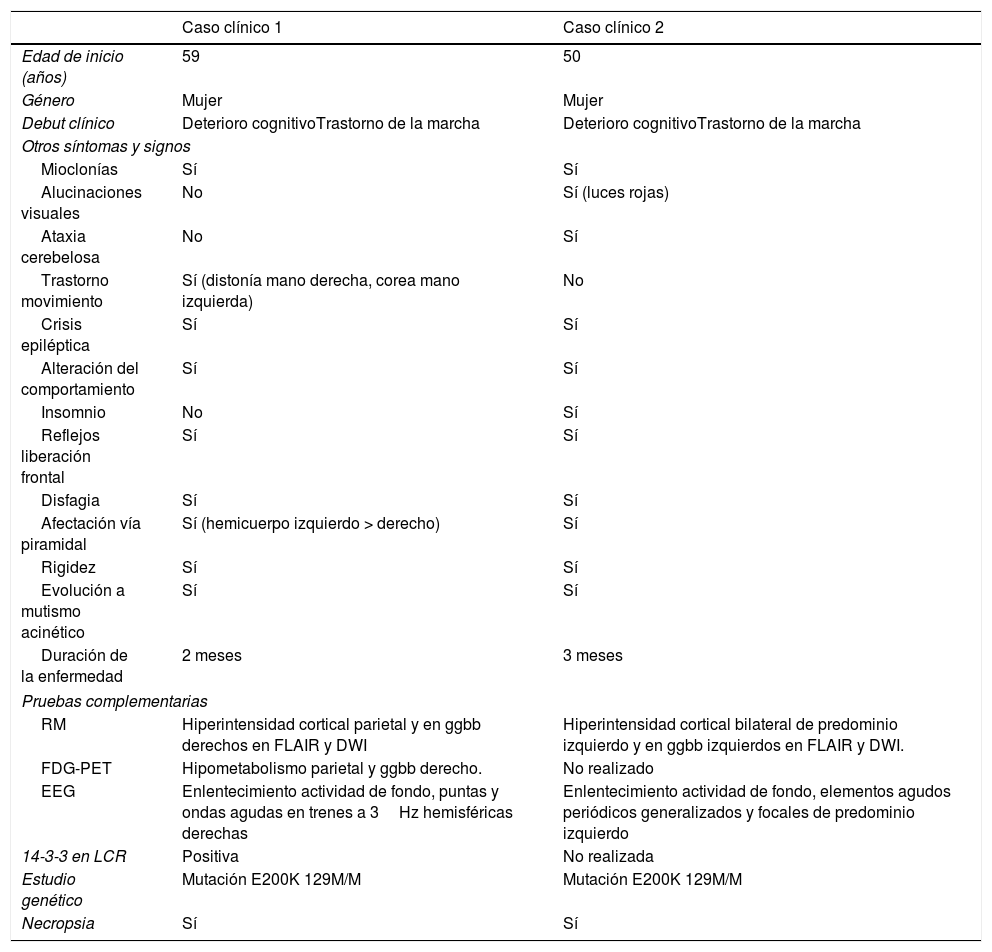
Características clínicas, hallazgos semiológicos y en las exploraciones complementarias de los casos clínicos presentados
| Caso clínico 1 | Caso clínico 2 | |
|---|---|---|
| Edad de inicio (años) | 59 | 50 |
| Género | Mujer | Mujer |
| Debut clínico | Deterioro cognitivoTrastorno de la marcha | Deterioro cognitivoTrastorno de la marcha |
| Otros síntomas y signos | ||
| Mioclonías | Sí | Sí |
| Alucinaciones visuales | No | Sí (luces rojas) |
| Ataxia cerebelosa | No | Sí |
| Trastorno movimiento | Sí (distonía mano derecha, corea mano izquierda) | No |
| Crisis epiléptica | Sí | Sí |
| Alteración del comportamiento | Sí | Sí |
| Insomnio | No | Sí |
| Reflejos liberación frontal | Sí | Sí |
| Disfagia | Sí | Sí |
| Afectación vía piramidal | Sí (hemicuerpo izquierdo > derecho) | Sí |
| Rigidez | Sí | Sí |
| Evolución a mutismo acinético | Sí | Sí |
| Duración de la enfermedad | 2 meses | 3 meses |
| Pruebas complementarias | ||
| RM | Hiperintensidad cortical parietal y en ggbb derechos en FLAIR y DWI | Hiperintensidad cortical bilateral de predominio izquierdo y en ggbb izquierdos en FLAIR y DWI. |
| FDG-PET | Hipometabolismo parietal y ggbb derecho. | No realizado |
| EEG | Enlentecimiento actividad de fondo, puntas y ondas agudas en trenes a 3Hz hemisféricas derechas | Enlentecimiento actividad de fondo, elementos agudos periódicos generalizados y focales de predominio izquierdo |
| 14-3-3 en LCR | Positiva | No realizada |
| Estudio genético | Mutación E200K 129M/M | Mutación E200K 129M/M |
| Necropsia | Sí | Sí |
DWI: diffusion-weighted imaging; EEG: electroencefalograma; FDG-PET: 18-fluorodesoxiglucosa tomografía por emisión de positrones; FLAIR: fluid-attenuated inversion recovery; ggbb: ganglios de la base; LCR: líquido cefalorraquídeo; RM: resonancia magnética.
 corresponden a Caso clínico 1. A) FDG-PET: hipometabolismo insular posterior y parietal derecho. B) RM DWI: hiperintensidad de predominio insular derecho. C) EEG vigilia (sistema 10/20): actividad lenta hemisférica bilateral y actividad epileptiforme focal parasagital derecha. Imágenes D-F) corresponden a Caso clínico 2. D) RM FLAIR y E) RM DWI: hiperintensidad cortical y de ganglios basales de predominio izquierdo. F) EEG vigilia (sistema 10/20): actividad theta-delta generalizada y elementos agudos tipo onda, punta-onda lenta de presentación periódica y predominio izquierdo.")
Imágenes A-C) corresponden a Caso clínico 1. A) FDG-PET: hipometabolismo insular posterior y parietal derecho. B) RM DWI: hiperintensidad de predominio insular derecho. C) EEG vigilia (sistema 10/20): actividad lenta hemisférica bilateral y actividad epileptiforme focal parasagital derecha. Imágenes D-F) corresponden a Caso clínico 2. D) RM FLAIR y E) RM DWI: hiperintensidad cortical y de ganglios basales de predominio izquierdo. F) EEG vigilia (sistema 10/20): actividad theta-delta generalizada y elementos agudos tipo onda, punta-onda lenta de presentación periódica y predominio izquierdo.
El padre de la paciente falleció con 63 años de un proceso neurodegenerativo rápidamente progresivo en 3 meses, no se hizo autopsia. Previa información y consejo, se realiza estudio genético a los familiares interesados. Dos de los cuatro hermanos (mujeres) de la paciente y uno de los dos hijos (varón) presenta la mutación E200K 129M/M. Uno de los hermanos (varón) rechaza estudiarse.
Caso clínico 2Mujer de 50 años, portadora de la mutación E200K 129M/M, hermana de la paciente anterior; presenta deterioro cognitivo progresivo y trastorno de la marcha en un mes. Fallece en estado de mutismo acinético a los 3 meses del inicio. Las características clínicas se resumen en la tabla 1. A los 60 días, la RM muestra hiperintensidad cortical en caudado y lenticular bilaterales con predominio hemisférico izquierdo en FLAIR y DWI (fig. 1D-E); el EEG registra elementos agudos periódicos generalizados y focales bilaterales con predominio hemisférico izquierdo (fig. 1F). Por deseo de los familiares y la paciente, no se realizaron otras exploraciones complementarias. En el estudio postmorten se observan cambios espongiformes en territorio cortical y subcortical de predominio izquierdo.
La notificación de ECJf en España es escasa y en solo 3 de los 12 casos reportados con origen genético se ha confirmado historia familiar3,4. En 2007, Morgado-Linares et al., presentaban otros 3 casos de una familia con la mutación E200K 129M/M5; la más frecuente en Europa6.
La RM de las dos pacientes mostró hiperintensidad cortical bilateral asimétrica y en ganglios de la base en secuencias FLAIR y DWI, hallazgos característicos de la ECJ esporádica (ECJe) y genética1. La afectación de los ganglios de la base es en todo caso más frecuente en pacientes con la mutación E200K, y se correlaciona con una menor supervivencia7,8.
En ningún EEG presentado se encontraron complejos agudos periódicos, característicos de la ECJ, siendo variable su incidencia en pacientes con mutación E200K (entre el 38% y 75%)2,9. En ambas pacientes encontramos concordancia entre el hemisferio afectado con mayor afectación cortical en la RM y lateralidad de los grafoelementos epileptiformes, aspectos conocidos en pacientes con la mutación E200K9.
El caso 1, al igual que otros casos de ECJe, presenta concordancia topográfica entre las zonas de hipometabolismo en el FDG-PET y los hallazgos radiológicos y neuropatológicos10. En este sentido, en el 51,5% de los casos de ECJe hay concordancia entre FDG-PET y alteraciones corticales en RM, y entre 80-34% entre FDG-PET y estudios posmorten11.
Las características clínicas de las dos pacientes son similares a las descritas en otros casos con la misma mutación, así como en el subtipo más frecuente de ECJe (MM1). La diferenciación clínica entre ECJe y ECJf es difícil. Las pruebas complementarias ayudan a caracterizar la enfermedad, y a diferenciar formas esporádicas de familiares. Se recomienda realizar siempre que sea posible, estudio genético al paciente y sus familiares para evitar catalogar casos familiares como esporádicos.
Como conclusión, se presentan dos casos de una misma familia con ECJf con la mutación E200K 129M/M, y al menos dos generaciones afectas. Se expone la correlación topográfica entre los hallazgos en FDG-PET, RM, EEG y neuropatología en uno de los casos; y entre RM, EEG y neuropatología en el otro.

