



Estudio descriptivo de epilepsias sintomáticas, según edad de inicio, controladas en una Unidad de Neuropediatría de referencia regional durante 3 años
Pacientes y métodosNiños con diagnóstico de epilepsia sintomática, controlados del 1 de enero del 2008 hasta el 31 de diciembre del 2010
ResultadosDe 4595 niños en el periodo de estudio, recibieron el diagnóstico de epilepsia 605 (13,17%), siendo 277 (45,79%) epilepsias sintomáticas. Entre los pacientes que iniciaron la epilepsia por debajo del año de vida predominan las de etiología sintomática (67,72%). Entre los que la iniciaron entre 1-3 años, fueron sintomáticas el 61,39%. En cuanto a su etiología, ha sido: encefalopatías prenatales (24,46% del total de epilepsias), encefalopatías perinatales (9,26%), encefalopatías posnatales (3,14%), encefalopatías metabólicas y degenerativas (1,98%), esclerosis mesial temporal (1,32%), síndromes neurocutáneos (2,64%), malformaciones vasculares (0,17%), cavernomas (0,17%) y tumores intracraneales (2,48%). Algunas etiologías inician sus manifestaciones epilépticas por debajo del año de vida, como el síndrome de Down, la lisencefalia genética, la infección congénita por citomegalovirus, la encefalopatía hipóxico-isquémica, las encefalopatías metabólicas o la esclerosis tuberosa
ConclusionesLa ausencia de una clasificación universalmente aceptada de los síndromes epilépticos dificulta comparaciones entre series. Sugerimos que todas las epilepsias son sintomáticas puesto que tienen causa, genética o adquirida. La edad de inicio orienta a determinadas etiologías. Una clasificación útil es la etiológica, con 2 grupos: un gran grupo con las etiologías establecidas o síndromes genéticos muy probables y otro de casos sin causa establecida, que con los avances en neuroimagen y genética cada vez será menor.
We conducted a descriptive study of symptomatic epilepsy by age at onset in a cohort of patients who were followed up at a neuropaediatric department of a reference hospital over a 3-year period
Patients and methodsWe included all children with epilepsy who were followed up from January 1, 2008 to December 31, 2010
ResultsOf the 4595 children seen during the study period, 605 (13.17%) were diagnosed with epilepsy; 277 (45.79%) of these had symptomatic epilepsy. Symptomatic epilepsy accounted for 67.72% and 61.39% of all epilepsies starting before one year of age, or between the ages of one and 3, respectively. The aetiologies of symptomatic epilepsy in our sample were: prenatal encephalopathies (24.46% of all epileptic patients), perinatal encephalopathies (9.26%), post-natal encephalopathies (3.14%), metabolic and degenerative encephalopathies (1.98%), mesial temporal sclerosis (1.32%), neurocutaneous syndromes (2.64%), vascular malformations (0.17%), cavernomas (0.17%), and intracranial tumours (2.48%). In some aetiologies, seizures begin before the age of one; these include Down syndrome, genetic lissencephaly, congenital cytomegalovirus infection, hypoxic-ischaemic encephalopathy, metabolic encephalopathies, and tuberous sclerosis.
ConclusionsThe lack of a universally accepted classification of epileptic syndromes makes it difficult to compare series from different studies. We suggest that all epilepsies are symptomatic because they have a cause, whether genetic or acquired. The age of onset may point to specific aetiologies. Classifying epilepsy by aetiology might be a useful approach. We could establish 2 groups: a large group including epileptic syndromes with known aetiologies or associated with genetic syndromes which are very likely to cause epilepsy, and another group including epileptic syndromes with no known cause. Thanks to the advances in neuroimaging and genetics, the latter group is expected to become increasingly smaller.
Las epilepsias incluyen un grupo de enfermedades del sistema nervioso central de etiología, pronóstico y tratamiento muy diversos1. Pueden ser la manifestación de diversos trastornos y en su etiología pueden intervenir tanto factores genéticos como adquiridos2,3.
Se denominan epilepsias sintomáticas a aquellas secundarias a alguna lesión cerebral subyacente, pudiendo presentarse en cualquier encefalopatía crónica, ya sea de origen prenatal, perinatal, posnatal o metabólico.
El objetivo de este trabajo es realizar un estudio descriptivo de las epilepsias sintomáticas, atendiendo a su edad de inicio y a su etiología, controladas en la Sección de Neuropediatría de un hospital de referencia regional durante un periodo de 3 años.
Material y métodosLa población a estudio está formada por todos los niños mayores de un mes de vida, diagnosticados de epilepsia sintomática que han sido valorados (por primera vez o en revisiones sucesivas) en la Unidad de Neuropediatría del Hospital Infantil Universitario Miguel Servet de Zaragoza durante un periodo de 3 años (del 1 de enero del 2008 al 31 de diciembre del 2010). La actividad asistencial desarrollada por esta unidad, desde su puesta en marcha en 1990, está recogida en una base de datos informatizada, con todos los datos de interés conocidos de cada uno de los pacientes valorados4-9. Los datos de cada paciente son actualizados cada vez que existe alguna incidencia reseñable en cuanto a evolución clínica, resultado de exámenes complementarios o cambio de tratamiento.
Se ha realizado un estudio descriptivo retrospectivo mediante la revisión de las historias clínicas de la población incluida, recogiendo variables epidemiológicas, características clínicas de la epilepsia, exámenes complementarios y datos evolutivos.
Se ha considerado epilepsia cuando se han dado al menos 2 crisis epilépticas espontáneas10. Se han excluido del estudio las convulsiones neonatales sin posterior epilepsia, las crisis afebriles aisladas, las convulsiones febriles y otras convulsiones provocadas o sintomáticas agudas.
Se ha considerado una epilepsia de etiología sintomática, cuando se ha detectado una lesión cerebral (estructural o metabólica) y que, además de las crisis convulsivas, se acompaña de otras manifestaciones neurológicas (si no existieran las convulsiones seguiría existiendo ese síndrome). Debido a su origen diverso, se ha elaborado una clasificación propia con grupos etiológicos para facilitar su estudio: 1) encefalopatías prenatales; 2) encefalopatías perinatales; 3) encefalopatías posnatales; 4) encefalopatías metabólicas y degenerativas; 5) esclerosis mesial temporal; 6) síndromes neurocutáneos; 7) malformaciones vasculares; 8) cavernomas; 9) tumores intracraneales, y 10) otras (tabla 1).
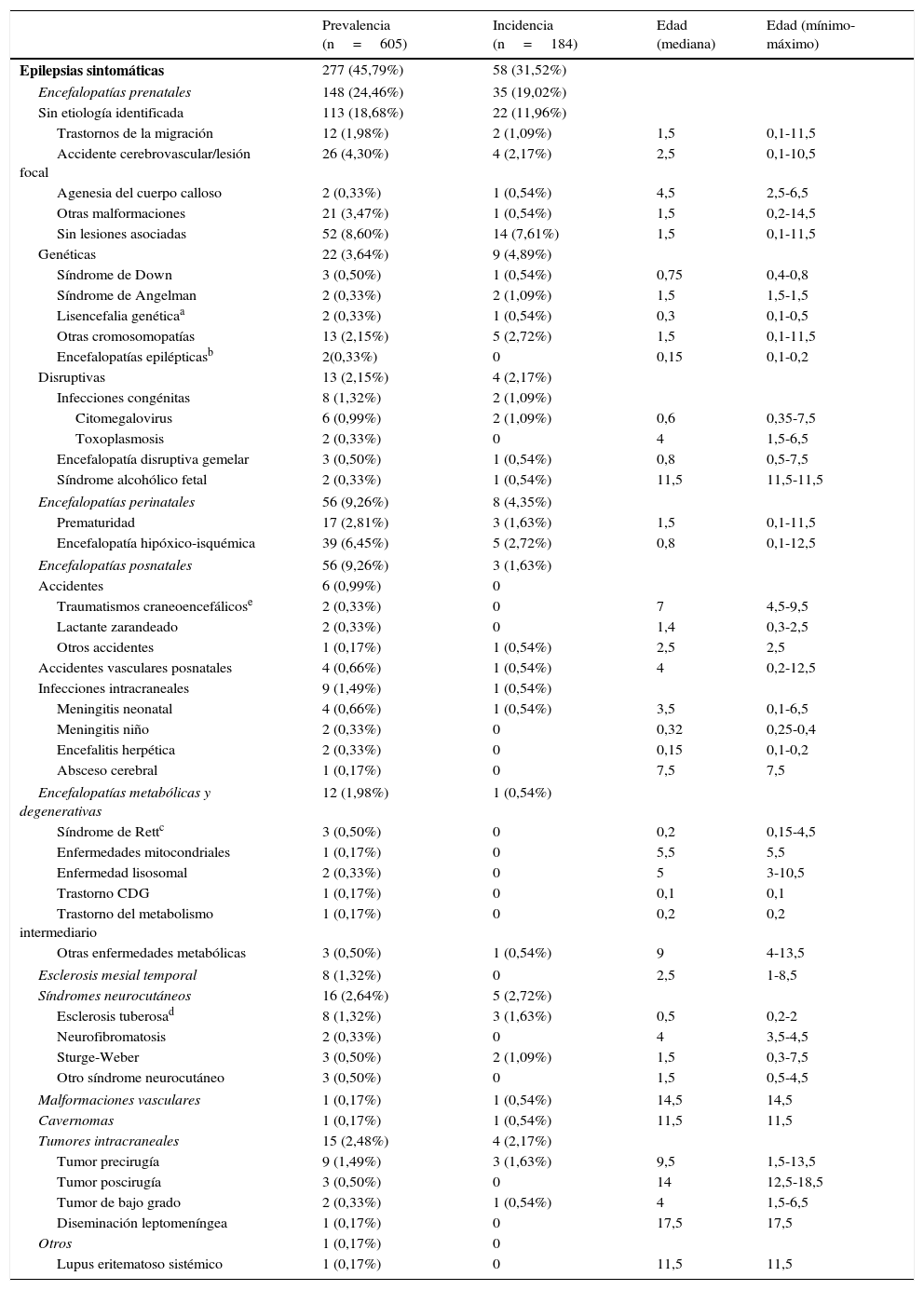
Datos de prevalencia, incidencia y edad de inicio (expresada en años) de las epilepsias sintomáticas (según grupos etiológicos) sobre el total de epilepsias de la unidad (N=605) durante el periodo a estudio (1 de enero del 2008 a 31 de diciembre del 2010)
| Prevalencia (n=605) | Incidencia (n=184) | Edad (mediana) | Edad (mínimo-máximo) | |
|---|---|---|---|---|
| Epilepsias sintomáticas | 277 (45,79%) | 58 (31,52%) | ||
| Encefalopatías prenatales | 148 (24,46%) | 35 (19,02%) | ||
| Sin etiología identificada | 113 (18,68%) | 22 (11,96%) | ||
| Trastornos de la migración | 12 (1,98%) | 2 (1,09%) | 1,5 | 0,1-11,5 |
| Accidente cerebrovascular/lesión focal | 26 (4,30%) | 4 (2,17%) | 2,5 | 0,1-10,5 |
| Agenesia del cuerpo calloso | 2 (0,33%) | 1 (0,54%) | 4,5 | 2,5-6,5 |
| Otras malformaciones | 21 (3,47%) | 1 (0,54%) | 1,5 | 0,2-14,5 |
| Sin lesiones asociadas | 52 (8,60%) | 14 (7,61%) | 1,5 | 0,1-11,5 |
| Genéticas | 22 (3,64%) | 9 (4,89%) | ||
| Síndrome de Down | 3 (0,50%) | 1 (0,54%) | 0,75 | 0,4-0,8 |
| Síndrome de Angelman | 2 (0,33%) | 2 (1,09%) | 1,5 | 1,5-1,5 |
| Lisencefalia genéticaa | 2 (0,33%) | 1 (0,54%) | 0,3 | 0,1-0,5 |
| Otras cromosomopatías | 13 (2,15%) | 5 (2,72%) | 1,5 | 0,1-11,5 |
| Encefalopatías epilépticasb | 2(0,33%) | 0 | 0,15 | 0,1-0,2 |
| Disruptivas | 13 (2,15%) | 4 (2,17%) | ||
| Infecciones congénitas | 8 (1,32%) | 2 (1,09%) | ||
| Citomegalovirus | 6 (0,99%) | 2 (1,09%) | 0,6 | 0,35-7,5 |
| Toxoplasmosis | 2 (0,33%) | 0 | 4 | 1,5-6,5 |
| Encefalopatía disruptiva gemelar | 3 (0,50%) | 1 (0,54%) | 0,8 | 0,5-7,5 |
| Síndrome alcohólico fetal | 2 (0,33%) | 1 (0,54%) | 11,5 | 11,5-11,5 |
| Encefalopatías perinatales | 56 (9,26%) | 8 (4,35%) | ||
| Prematuridad | 17 (2,81%) | 3 (1,63%) | 1,5 | 0,1-11,5 |
| Encefalopatía hipóxico-isquémica | 39 (6,45%) | 5 (2,72%) | 0,8 | 0,1-12,5 |
| Encefalopatías posnatales | 56 (9,26%) | 3 (1,63%) | ||
| Accidentes | 6 (0,99%) | 0 | ||
| Traumatismos craneoencefálicose | 2 (0,33%) | 0 | 7 | 4,5-9,5 |
| Lactante zarandeado | 2 (0,33%) | 0 | 1,4 | 0,3-2,5 |
| Otros accidentes | 1 (0,17%) | 1 (0,54%) | 2,5 | 2,5 |
| Accidentes vasculares posnatales | 4 (0,66%) | 1 (0,54%) | 4 | 0,2-12,5 |
| Infecciones intracraneales | 9 (1,49%) | 1 (0,54%) | ||
| Meningitis neonatal | 4 (0,66%) | 1 (0,54%) | 3,5 | 0,1-6,5 |
| Meningitis niño | 2 (0,33%) | 0 | 0,32 | 0,25-0,4 |
| Encefalitis herpética | 2 (0,33%) | 0 | 0,15 | 0,1-0,2 |
| Absceso cerebral | 1 (0,17%) | 0 | 7,5 | 7,5 |
| Encefalopatías metabólicas y degenerativas | 12 (1,98%) | 1 (0,54%) | ||
| Síndrome de Rettc | 3 (0,50%) | 0 | 0,2 | 0,15-4,5 |
| Enfermedades mitocondriales | 1 (0,17%) | 0 | 5,5 | 5,5 |
| Enfermedad lisosomal | 2 (0,33%) | 0 | 5 | 3-10,5 |
| Trastorno CDG | 1 (0,17%) | 0 | 0,1 | 0,1 |
| Trastorno del metabolismo intermediario | 1 (0,17%) | 0 | 0,2 | 0,2 |
| Otras enfermedades metabólicas | 3 (0,50%) | 1 (0,54%) | 9 | 4-13,5 |
| Esclerosis mesial temporal | 8 (1,32%) | 0 | 2,5 | 1-8,5 |
| Síndromes neurocutáneos | 16 (2,64%) | 5 (2,72%) | ||
| Esclerosis tuberosad | 8 (1,32%) | 3 (1,63%) | 0,5 | 0,2-2 |
| Neurofibromatosis | 2 (0,33%) | 0 | 4 | 3,5-4,5 |
| Sturge-Weber | 3 (0,50%) | 2 (1,09%) | 1,5 | 0,3-7,5 |
| Otro síndrome neurocutáneo | 3 (0,50%) | 0 | 1,5 | 0,5-4,5 |
| Malformaciones vasculares | 1 (0,17%) | 1 (0,54%) | 14,5 | 14,5 |
| Cavernomas | 1 (0,17%) | 1 (0,54%) | 11,5 | 11,5 |
| Tumores intracraneales | 15 (2,48%) | 4 (2,17%) | ||
| Tumor precirugía | 9 (1,49%) | 3 (1,63%) | 9,5 | 1,5-13,5 |
| Tumor poscirugía | 3 (0,50%) | 0 | 14 | 12,5-18,5 |
| Tumor de bajo grado | 2 (0,33%) | 1 (0,54%) | 4 | 1,5-6,5 |
| Diseminación leptomeníngea | 1 (0,17%) | 0 | 17,5 | 17,5 |
| Otros | 1 (0,17%) | 0 | ||
| Lupus eritematoso sistémico | 1 (0,17%) | 0 | 11,5 | 11,5 |
El término encefalopatía se ha utilizado obedeciendo a su significado etimológico de padecimiento encefálico, independientemente de su carácter difuso o localizado y de las repercusiones clínicas. Se han considerado encefalopatías posnatales a las secundarias a infecciones del sistema nervioso central, traumatismos y accidentes cerebrovasculares posnatales. El diagnóstico de encefalopatía prenatal se ha establecido incluyendo criterios clínicos y/o de neuroimagen. Así, apoyan el origen prenatal de una encefalopatía datos como la existencia de polihidramnios, rasgos dismórficos faciales y malformaciones extraneurológicas asociadas, y la ausencia de evidencia de noxa perinatal o posnatal. La identificación por neuroimagen de agenesia del cuerpo calloso, trastornos de la migración o de otras anomalías malformativas es diagnóstica de encefalopatía prenatal.
ResultadosEn el momento del estudio, la base de datos de la Unidad de Neuropediatría tenía 15.808 pacientes registrados. Durante el periodo de estudio han sido atendidos en la Unidad un total de 4.595 pacientes. De ellos, en 1.654 pacientes el motivo de consulta fue un trastorno paroxístico (35,99%) y 605 fueron diagnosticados de epilepsia (13,17% del total de pacientes, 36,58% de los trastornos paroxísticos). La etiología de la epilepsia ha sido considerada como sintomática en 277 casos (45,79%), siendo el 54,71% varones y el 45,29% mujeres. Durante los 3 años de estudio se diagnosticaron 184 casos nuevos de epilepsia, habiéndose considerado 58 (31,52%) como epilepsias sintomáticas. El tiempo medio de seguimiento del total de epilepsias ha sido de 6,21 años, siendo el de las sintomáticas de 8,13 años.
La edad media de inicio del total de las epilepsias fue de 4,78 años, siendo la de las sintomáticas de 3,53 años. La máxima incidencia ha sido en el primer año de vida: el 26,12% del total de las epilepsias. Por grupos de edad, en los pacientes que iniciaron la epilepsia por debajo del año de vida predominan las de etiología sintomática (67,72%), al igual que en las epilepsias de inicio entre 1-3 años (61,39%). Clínicamente, el 71,74% de las epilepsias sintomáticas se presentaron con crisis convulsivas focales o parciales, el 13,41% como crisis convulsivas generalizadas, el 12,68% como espasmos epilépticos y el resto como crisis indeterminadas (habiendo presentado estatus convulsivo el 4,71%). El 2,54% de nuestras epilepsias sintomáticas tenían antecedentes familiares de epilepsia, el 15,22% había presentado convulsiones en el periodo neonatal y el 10,14% convulsiones febriles.
La tabla 1 expone los datos de incidencia, prevalencia y edad de inicio de las epilepsias sintomáticas, según grupos etiológicos.
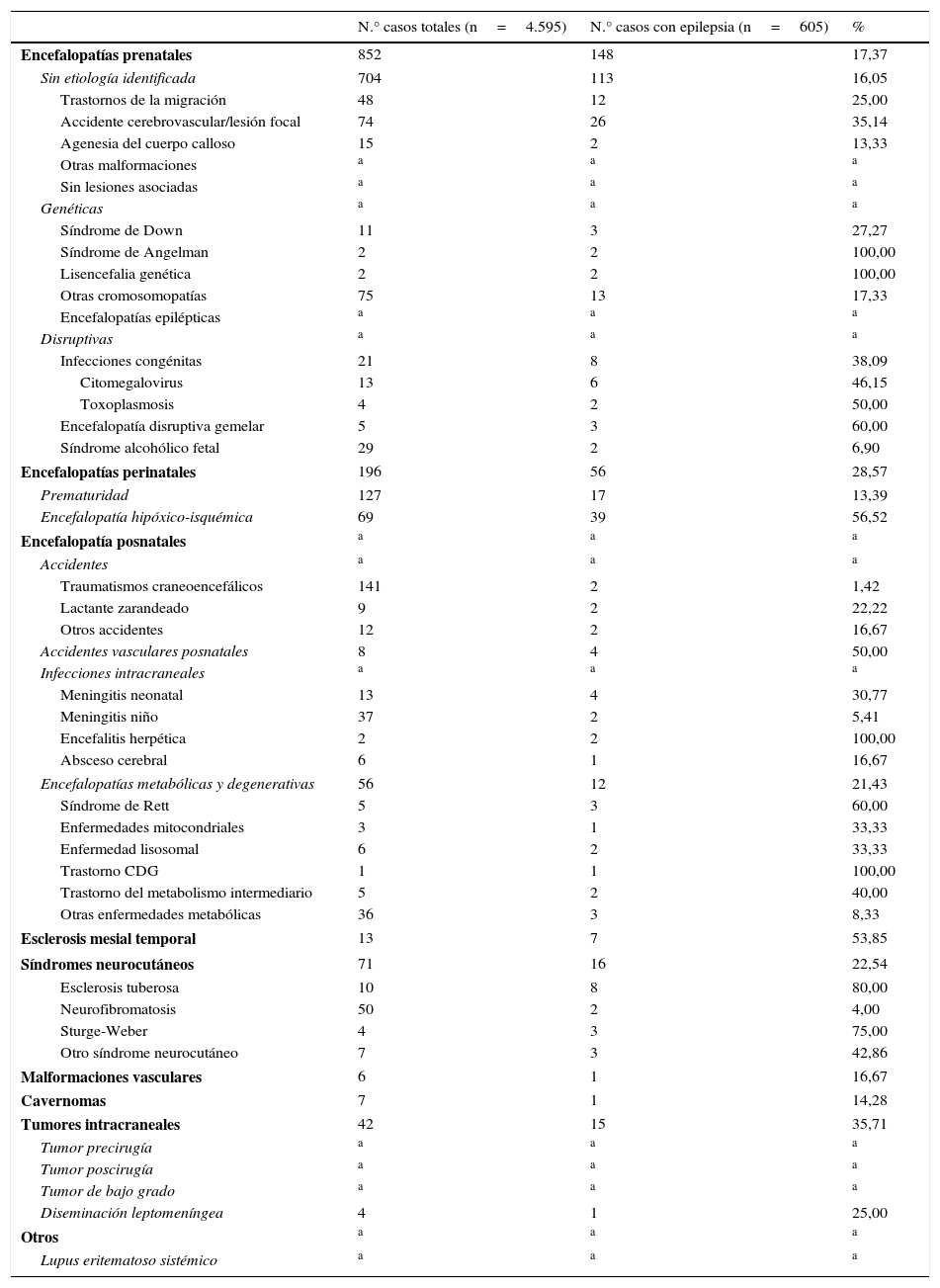
La tabla 2 recoge la frecuencia de las epilepsias sintomáticas respecto al total de casos de la base de datos de neuropediatría durante el periodo de estudio de cada una de las patologías que asocian epilepsia.
Frecuencia de epilepsia en cada una de las patologías señaladas en las epilepsias sintomáticas del total de los 4.595 pacientes valorados en la Unidad de Neuropediatría durante el periodo de estudio (1 de enero del 2008 a 31 de diciembre del 2010)
| N.° casos totales (n=4.595) | N.° casos con epilepsia (n=605) | % | |
|---|---|---|---|
| Encefalopatías prenatales | 852 | 148 | 17,37 |
| Sin etiología identificada | 704 | 113 | 16,05 |
| Trastornos de la migración | 48 | 12 | 25,00 |
| Accidente cerebrovascular/lesión focal | 74 | 26 | 35,14 |
| Agenesia del cuerpo calloso | 15 | 2 | 13,33 |
| Otras malformaciones | a | a | a |
| Sin lesiones asociadas | a | a | a |
| Genéticas | a | a | a |
| Síndrome de Down | 11 | 3 | 27,27 |
| Síndrome de Angelman | 2 | 2 | 100,00 |
| Lisencefalia genética | 2 | 2 | 100,00 |
| Otras cromosomopatías | 75 | 13 | 17,33 |
| Encefalopatías epilépticas | a | a | a |
| Disruptivas | a | a | a |
| Infecciones congénitas | 21 | 8 | 38,09 |
| Citomegalovirus | 13 | 6 | 46,15 |
| Toxoplasmosis | 4 | 2 | 50,00 |
| Encefalopatía disruptiva gemelar | 5 | 3 | 60,00 |
| Síndrome alcohólico fetal | 29 | 2 | 6,90 |
| Encefalopatías perinatales | 196 | 56 | 28,57 |
| Prematuridad | 127 | 17 | 13,39 |
| Encefalopatía hipóxico-isquémica | 69 | 39 | 56,52 |
| Encefalopatía posnatales | a | a | a |
| Accidentes | a | a | a |
| Traumatismos craneoencefálicos | 141 | 2 | 1,42 |
| Lactante zarandeado | 9 | 2 | 22,22 |
| Otros accidentes | 12 | 2 | 16,67 |
| Accidentes vasculares posnatales | 8 | 4 | 50,00 |
| Infecciones intracraneales | a | a | a |
| Meningitis neonatal | 13 | 4 | 30,77 |
| Meningitis niño | 37 | 2 | 5,41 |
| Encefalitis herpética | 2 | 2 | 100,00 |
| Absceso cerebral | 6 | 1 | 16,67 |
| Encefalopatías metabólicas y degenerativas | 56 | 12 | 21,43 |
| Síndrome de Rett | 5 | 3 | 60,00 |
| Enfermedades mitocondriales | 3 | 1 | 33,33 |
| Enfermedad lisosomal | 6 | 2 | 33,33 |
| Trastorno CDG | 1 | 1 | 100,00 |
| Trastorno del metabolismo intermediario | 5 | 2 | 40,00 |
| Otras enfermedades metabólicas | 36 | 3 | 8,33 |
| Esclerosis mesial temporal | 13 | 7 | 53,85 |
| Síndromes neurocutáneos | 71 | 16 | 22,54 |
| Esclerosis tuberosa | 10 | 8 | 80,00 |
| Neurofibromatosis | 50 | 2 | 4,00 |
| Sturge-Weber | 4 | 3 | 75,00 |
| Otro síndrome neurocutáneo | 7 | 3 | 42,86 |
| Malformaciones vasculares | 6 | 1 | 16,67 |
| Cavernomas | 7 | 1 | 14,28 |
| Tumores intracraneales | 42 | 15 | 35,71 |
| Tumor precirugía | a | a | a |
| Tumor poscirugía | a | a | a |
| Tumor de bajo grado | a | a | a |
| Diseminación leptomeníngea | 4 | 1 | 25,00 |
| Otros | a | a | a |
| Lupus eritematoso sistémico | a | a | a |
A pesar de los avances en epileptología, resulta difícil realizar estudios epidemiológicos debido a problemas metodológicos y a la falta de uniformidad de criterios, existiendo por ello grandes diferencias estadísticas entre los diferentes trabajos publicados2,3,11,12. La proporción de epilepsias sintomáticas en series comparables oscila entre el 18,1 y el 50%3,13-17, siendo en nuestro caso del 45,79% de prevalencia y del 31,52% de incidencia. En nuestra serie, como en la mayoría de las publicaciones, se aprecia un discreto predominio de varones frente a las mujeres18-20.
Los síndromes epilépticos son dependientes de la edad y sus características clínicas y electroencefalográficas dependen del grado de maduración cerebral21-23, como se ve en nuestro estudio. Así, en función de la edad son más frecuentes unas etiologías y tipos de epilepsias que otros.
Las epilepsias sintomáticas comienzan a edades más precoces: el 67,72% el primer año de vida. La etiología de las crisis infantiles precoces está dominada por la presencia de lesiones cerebrales. Muchas de las lesiones responsables de epilepsia infantil precoz tienen un origen prenatal y perinatal24. Un lactante que inicia la epilepsia en el primer mes de vida tiene muchas posibilidades de que sea una epilepsia sintomática, en nuestro caso, el 89,74%. En nuestra experiencia, en este periodo de edad las etiologías de las epilepsias son fundamentalmente por encefalopatías perinatales (43,59%) y encefalopatías prenatales (38,46%). En los lactantes de 1 a 3 meses, también es más probable que sea de etiología sintomática, el 66,67% en nuestro estudio, siendo lo más frecuente encefalopatías prenatales (33,33%). En el grupo de 3 a 12 meses, igualmente las epilepsias sintomáticas son las más frecuentes, el 58,42% según nuestros datos.
Nuestra serie muestra que existen ciertas etiologías que generalmente inician sus manifestaciones epilépticas por debajo del año de vida, como son el síndrome de Down, la lisencefalia genética, la infección congénita por citomegalovirus, la encefalopatía hipóxico-isquémica, las encefalopatías metabólicas o la esclerosis tuberosa (tabla 1).
Los pacientes que inician su epilepsia entre 1 y 3 años también tienen más posibilidades de que sean de etiología sintomática; según nuestros datos, el 61,39%: encefalopatías prenatales y perinatales, y casos de esclerosis mesial temporal.
Por encima de los 3 años de edad, la etiología sintomática de la epilepsia es menos frecuente (31,21%): infecciones intracraneales, accidentes, tumores intracraneales o malformaciones vasculares fundamentalmente.
En la muestra total, el 53,42% de las epilepsias sintomáticas (correspondiente al 24,46% del total de las epilepsias) se deben a encefalopatías prenatales. Estas son aquellas que se producen antes del nacimiento del niño pudiendo ser disruptivas (problemas vasculares, tóxicos, infecciones, etc.) o estar genéticamente determinadas. En muchas ocasiones, una adecuada interpretación de hallazgos de neuroimagen (malformaciones cerebrales que son dependientes del momento gestacional en que se producen) y datos clínicos nos permite establecer su origen prenatal, pero no permiten identificar su diagnóstico etiológico25,26. En nuestra experiencia, no se ha llegado a identificar la etiología en 113 pacientes: el 40,79% de todas las epilepsias sintomáticas y el 76,35% de las epilepsias secundarias a encefalopatías prenatales.
Dentro de las malformaciones cerebrales, destacan los trastornos de la migración/proliferación y otros trastornos del desarrollo cortical, que están muy a menudo asociadas a crisis epilépticas y, además, frecuentemente refractarias al tratamiento médico27. Estas son entidades con gran variabilidad de presentación (según en la etapa de desarrollo cerebral en que se produzcan y la propia etiología), lo que marcará su funcionalidad y el grado de epileptogenicidad28. En el proceso de migración intervienen multitud de factores, tanto genéticos como ambientales; así, puede verse alterado por fenómenos hipoxicoisquémicos, infecciosos (como el citomegalovirus), drogas, tóxicos, venenos o irradiaciones29. En nuestra muestra se ha recogido a 12 niños epilépticos con algún trastorno de la migración o displasia cortical sin haber identificado su etiología.
Las encefalopatías prenatales en nuestra experiencia tienen una incidencia de epilepsia del 17,37%, pero, dentro de ellas, en el síndrome de Angelman o las lisencefalias genéticas es del 100%, la infección congénita por toxoplasma del 50% y por citomegalovirus del 46,15%.
En los últimos años, gracias a la mejora en la atención obstétrica y los avances en neonatología, se ha conseguido que aumente la supervivencia de los recién nacidos prematuros y los recién nacidos con asfixia perinatal, aunque persisten secuelas neurológicas importantes, como es la epilepsia30,31. En nuestra serie suponen el 9,26% de todas las epilepsias y el 20,22% de las sintomáticas, pero teniendo una mayor incidencia de epilepsia las encefalopatías hipóxico-isquémicas (56,52%) que los casos de prematuridad (13,39%), probablemente debido a que en los primeros ya existe una lesión implícita en su diagnóstico y en los casos de prematuridad, debido a la mejoría en los cuidados neonatales, han disminuido considerablemente la aparición de lesiones cerebrales, como la leucomalacia periventricular.
Los traumatismos craneoencefálicos suelen considerarse una causa frecuente de epilepsia; se estima que la incidencia de epilepsia postraumática tardía en los casos graves oscila entre el 1 y el 57%32, existiendo un mayor riesgo en traumatismos abiertos, si hubo hematoma intracraneal o si presentó convulsiones en la primera semana33,34. En nuestra muestra solo se han encontrado 2 casos (0,72% de las epilepsias sintomáticas): los 2 presentaron hematomas intracraneales, uno fue un traumatismo abierto y ninguno presentó convulsiones en la primera semana.
La esclerosis mesial temporal es otra causa posible de epilepsia, si bien su patogenia no está del todo aclarada. Se han descrito diversos factores desencadenantes de la lesión del hipocampo (pérdida neuronal y posterior esclerosis), como traumatismos craneales, infartos perinatales, infecciones intracraneales35,36. Parece que además podría existir una predisposición genética que favorezca la aparición de este síndrome35,37. En la muestra se han recogido 7 casos (el 1,16% del total de las epilepsias y el 2,52% de las sintomáticas) en los que no se ha identificado la etiología de la lesión y 2 pacientes con esclerosis mesial e infección congénita por citomegalovirus. En nuestra serie, el 53,85% de los pacientes con esclerosis mesial temporal tienen epilepsia.
Globalmente, se da epilepsia en el 22,54% de nuestros casos de síndromes neurocutáneos. Dentro de los síndromes neurocutáneos, a pesar de que la neurofibromatosis tipo 1 es la más frecuente (en nuestra casuística supone el 70,42% de ellos), se estima que la prevalencia de epilepsia es menor a la de los otros síndromes neurocutáneos, considerada entre el 3-8%38,39, como en nuestra experiencia, el 4% (2 casos de los 50 controlados) tienen epilepsia. Por el contrario, los pacientes con esclerosis tuberosa, aunque son menos numerosos, precisan más visitas en las consultas de Neuropediatría que los de neurofibromatosis por las altas tasas de epilepsia, que oscilan entre el 78 y el 95%38,40,41, como en nuestra serie, que es del 80% (8 casos de los 10 que se han controlado presentan epilepsia); además, con frecuencia de inicio en el primer año de vida. Igualmente, en el síndrome de Sturge-Weber, la tasa de epilepsia se estima en el 80% de los casos de afectación unilateral y hasta el 93% si hay afectación bilateral, similar a nuestros datos, con datos de epilepsia en el 75% de los casos (3 de los 4 pacientes controlados durante el periodo).
Las crisis convulsivas pueden ser a menudo el primer síntoma de un tumor cerebral (20-50%), aunque pueden presentar otros síntomas neurológicos asociados42,43. Su incidencia estimada como causa de epilepsia en edades pediátricas es del 0,2-6%43,44, siendo en nuestra serie del 2,48% (suponiendo el 5,42% de las epilepsias sintomáticas). De nuestros 15 casos de tumores cerebrales, en 6 las convulsiones fueron el síntoma de presentación (40%) y el resto presentó las crisis durante su evolución.
En el abordaje de la epilepsia en la infancia, la edad de inicio es determinante; en niños mayores en general puede ser suficiente con los estudios de neuroimagen y electroencefalograma, y en lactantes pueden ser necesarios amplios estudios, metabólicos y genéticos21. En nuestra serie, no hemos identificado ninguna epilepsia de origen autoinmune, pero cada vez se conocen más casos de ese origen y es necesario planteárselo para el adecuado, aunque complejo, diagnóstico y manejo terapéutico45-47. En las epilepsias precoces, y en particular cuando las crisis empiezan entre 1 y 4 meses, es mucho más frecuente que la causa sea un grave problema cerebral con mala respuesta al tratamiento antiepiléptico y mal pronóstico neurológico y del desarrollo48. Raramente pueden deberse a enfermedades metabólicas hereditarias, algunas de las cuales pueden tener tratamiento específico (con vitaminas o dieta cetógena) y que no responden a los fármacos antiepilépticos. Dados la importante preocupación en cuanto al pronóstico y el riesgo de repetición (al tratarse con frecuencia de un problema genético), junto a las opciones, aunque poco frecuentes, de respuesta a tratamiento específico, es necesario un protocolo diagnóstico-terapéutico que permita, en el caso que sea posible, establecer un tratamiento precoz e identificar la causa y que contemple el tratamiento con vitaminas, siempre tras la recogida de muestras biológicas. En epilepsias focales refractarias a cualquier edad debe insistirse en las técnicas de neuroimagen y neuroimagen funcional para descartar lesiones potencialmente quirúrgicas, puesto que la lesionectomía puede ser curativa49,50.
La ausencia de una clasificación universalmente aceptada de los síndromes epilépticos51,52 dificulta comparaciones entre series, empezando por la terminología. Las encefalopatías epilépticas con mutación identificada, como nuestros casos de STXBP1 y CDKL5, y el síndrome de Dravet (habitualmente considerado idiopático, por lo que no lo hemos incluido en la serie) podrían clasificarse como sintomáticos (encefalopatía genéticamente determinada con disfunción del neurodesarrollo no necesariamente secundaria a la epilepsia) y como idiopáticos (genéticamente determinados y la epilepsia es nuclear). Las encefalopatías prenatales no disruptivas, los síndromes neurocutáneos, las enfermedades metabólicas degenerativas y muchos casos de malformaciones vasculares, cavernomas, tumores cerebrales y esclerosis mesial temporal están genéticamente determinados. En nuestra opinión, todas las epilepsias son sintomáticas puesto que tienen una etiología determinada, sea genética o adquirida. La edad de inicio resulta ser orientativa para el diagnóstico etiológico en determinadas ocasiones.
Quizás una clasificación etiológica podría ser más útil al clasificar la epilepsia en 2 grupos: un gran grupo con las etiologías establecidas o síndromes genéticos muy probables y otro de casos sin causa establecida, que con los avances en neuroimagen y genética cada vez será menor.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

