



La paraplejia espástica tipo 7 (SPG7) (MIM # 607259), es una enfermedad autosómica recesiva (AR) causada por variantes en el gen SPG7 que codifica la paraplegina, una metaloproteasa mitocondrial1. Representa aproximadamente el 5-12% de las formas autosómicas recesivas y hasta el 7% de los casos esporádicos del adulto. Posteriormente se ha relacionado con un complejo sintomático asociado a ataxia cerebelosa2, pudiendo ser esta la manifestación clínica más relevante. Sin embargo, las variaciones fenotípicas van más allá, describiéndose casos en la literatura de oftalmoplejía externa progresiva crónica3, esclerosis lateral primaria4 y parkinsonismo5, entre otros.
Se han descrito más de 131 variantes patogénicas (HGMD visitado el 14/07/2019). Reportamos los casos de 3 hermanas de una familia española con un fenotipo de ataxia y espasticidad, y portadoras de 2 variantes en un solo nucleótido (SNV) en el gen SPG7 de significado incierto hasta el momento.
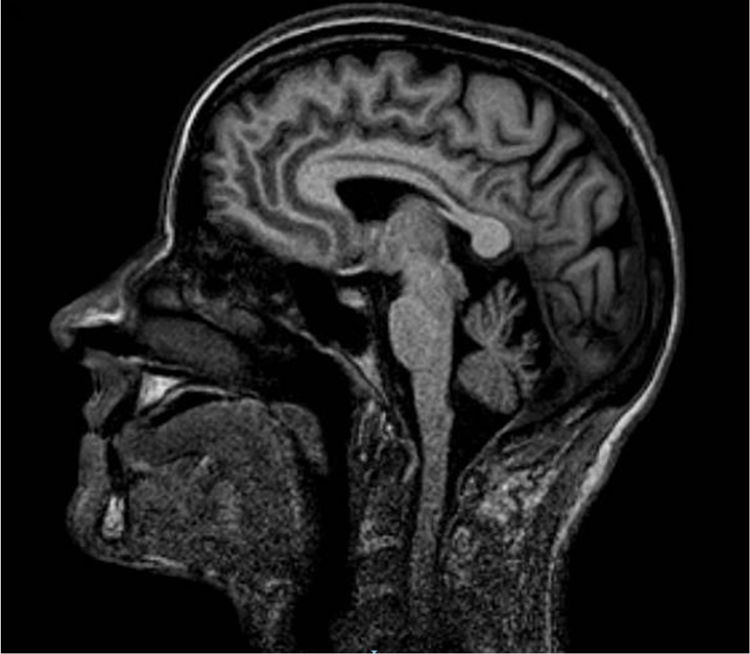
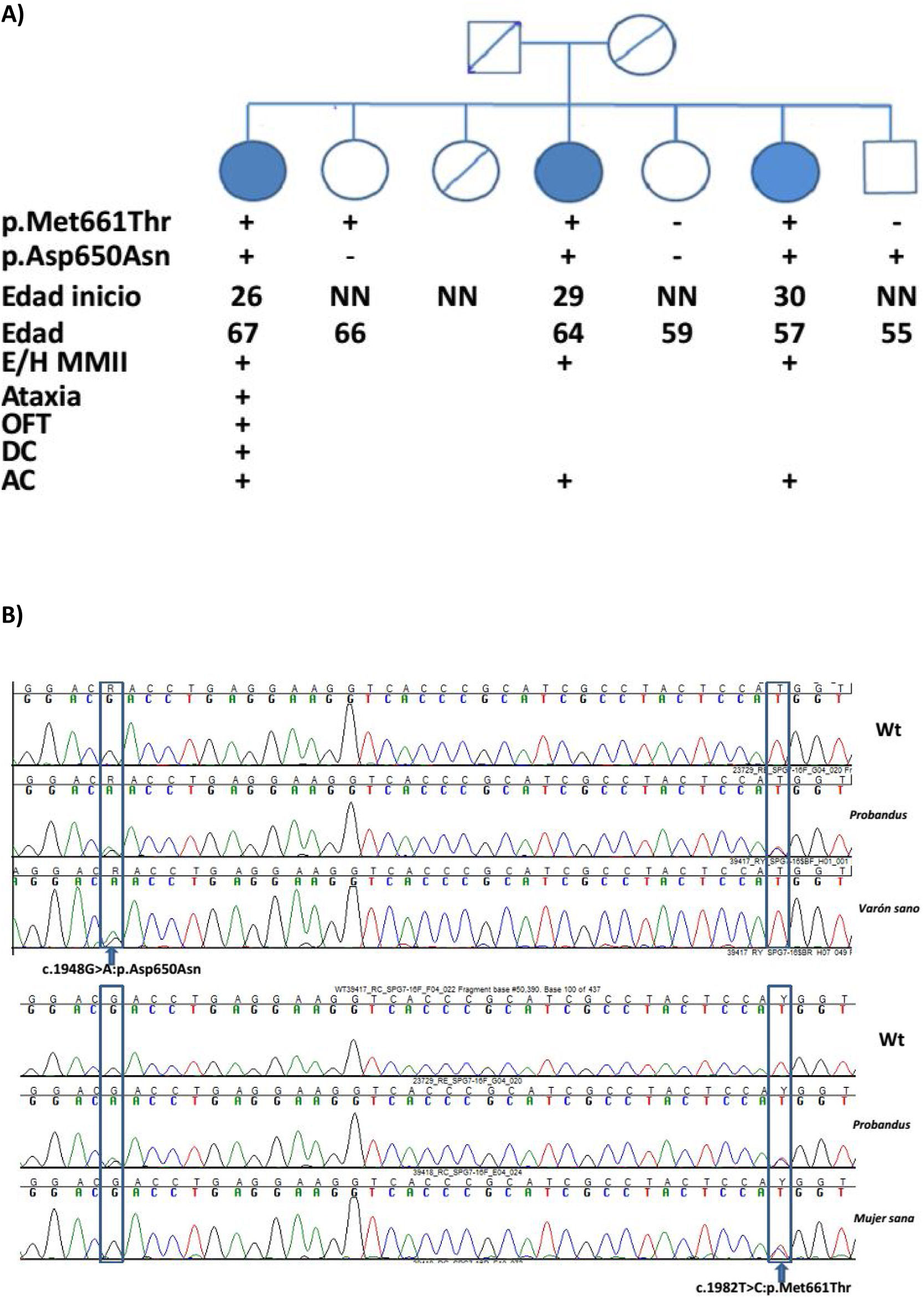
Estudiamos una familia de 7 hermanos de padres no consanguíneos, sin antecedentes familiares de enfermedad neurológica. Tres de las hermanas referían dificultad progresiva en la marcha con inestabilidad y disartria de inicio en la tercera década de la vida. En la exploración clínica tras 20 años de evolución destacaba: nistagmo multidireccional, disartria, hiperreflexia global con reflejo cutáneo plantar extensor bilateral, espasticidad marcada en las extremidades inferiores requiriendo soporte bilateral para caminar. La de mayor edad asociaba, además: deterioro cognitivo, oftalmoparesia, dismetría en extremidades superiores y aumento de la base para caminar. Todas mostraron atrofia cerebelosa en resonancia (fig. 1). El resto de los hermanos no estaban afectados (fig. 2A).

 Pedigree. AC: atrofia cerebral; DC: deterioro cognitivo; E/HMMII: espasticidad/hiperreflexia en miembros inferiores; NN: neurológicamente normal; OFP: oftalmoparesia. +: presente; −: ausente. B) Secuencia Sanger de SPG7. Mutación en heterocigosis indicada con flechas.")
Mutación SPG7 y correlación clínica. A) Pedigree. AC: atrofia cerebral; DC: deterioro cognitivo; E/HMMII: espasticidad/hiperreflexia en miembros inferiores; NN: neurológicamente normal; OFP: oftalmoparesia. +: presente; −: ausente. B) Secuencia Sanger de SPG7. Mutación en heterocigosis indicada con flechas.
Tras descartar causas secundarias y las mutaciones más frecuentes de ataxia y paraparesia espástica en el probando, se llevó a cabo el estudio genético para el gen SPG7. Al detectar 2 variantes missense, el estudio se extendió al resto de hermanos.
Se amplificó el ADN genómico extraído de muestras de sangre periférica mediante PCR y se realizó secuenciación convencional en ABI 3730® (Applied Biosystems, Foster City, CA, EE. UU.). El diseño cubría todos los exones codificantes, así como todos los límites exón-intrón. También se realizó un análisis de pequeñas deleciones/inserciones y mutaciones puntuales en la región codificante y los sitios splicing del gen SPG7.
Se detectaron 2 variantes missense en heterocigosis compuesta (NM_003119.3:c.1982T>C:p.Met661Thr y NM_003119.3:c.1948G>A:p.Asp650Asn) en las 3 hermanas afectas. Dos hermanos no afectos eran portadores alternativamente de cada una de las variantes y una tercera no presentaba ninguna de ellas (fig. 2B).
La interpretación genético-clínica de las variantes se llevó a cabo estudiando las frecuencias poblacionales, algoritmos de conservación (GERP, Phylophen, SiPhy) y de predicción de impacto funcional (SIFT, Polyphen, Mutation Taster, Mutation Assesor, LRT, CADD, Dann) en cada una de las variantes. Se revisó la información sobre variantes genéticas presentes en el gen SPG7 previamente asociadas a la enfermedad publicadas en la literatura, así como a través de distintas fuentes online (OMIM®, GeneReviews®, ClinVar, HGMD™ mutation database). También se estudió la segregación familiar, aunque no había muestra disponible de ninguno de los progenitores.
Las predicciones in silico y los criterios clínicos y genéticos adicionales estudiados respaldan la posible patogenicidad de ambas variantes. La variante p.Met661Thr no estaba presente en las bases de datos de frecuencias poblacionales, y se había detectado en heterocigosidad compuesta en un paciente con sospecha de paraparesia espástica pura6 y p.Asp650Asn presenta una frecuencia poblacional del 0,001%, y se había detectado previamente en un caso esporádico de un paciente con ataxia como rasgo primario y afectación de neurona motora superior7. En ambos casos, todos los algoritmos de conservación y predicción de patogenicidad señalaron impacto funcional en la proteína.
La familia descrita muestra un cuadro clínico característico, aunque con algunas salvedades. Hay que destacar que la paciente con mayor evolución de la enfermedad, además de presentar un mayor componente atáxico, manifiesta síntomas menos comunes como son la oftalmoparesia y el deterioro cognitivo.
Estas diferencias fenotípicas podrían deberse a una sumación de síntomas durante la progresión de la enfermedad, aunque no disponemos de un seguimiento prospectivo.
Ambas variantes han sido clasificadas de significado incierto, descritos en tan solo 2 casos en la literatura. Los datos clínico-genéticos que reportamos avalan la hipótesis de su papel patógeno. No obstante, se requieren más estudios para evaluar con precisión la patogenicidad.
Hasta donde sabemos, este es el primer estudio donde ambas variantes se recogen juntas en una familia con ataxia espástica. Aunque la muestra de los padres no estaba disponible, la segregación de cada una de las variantes en diferentes hermanos afectos y no afectos demuestra que cada una de ellas proviene de un padre diferente.

