



Cerebrospinal fluid (CSF) from amyotrophic lateral sclerosis (ALS) patients induces cytotoxic effects in in vitro cultured motor neurons.
Materials and methodsWe selected CSF with previously reported cytotoxic effects from 32 ALS patients. Twenty-eight adult male rats were intracerebroventricularly implanted with osmotic mini-pumps and divided into 3 groups: 9 rats injected with CSF from non-ALS patients, 15 rats injected with cytotoxic ALS-CSF, and 4 rats injected with a physiological saline solution. CSF was intracerebroventricularly and continuously infused for periods of 20 or 43days after implantation. We conducted clinical assessments and electromyographic examinations, and histological analyses were conducted in rats euthanised 20, 45, and 82days after surgery.
ResultsImmunohistochemical studies revealed tissue damage with similar characteristics to those found in the sporadic forms of ALS, such as overexpression of cystatin C, transferrin, and TDP-43 protein in the cytoplasm. The earliest changes observed seemed to play a protective role due to the overexpression of peripherin, AKTpan, AKTphospho, and metallothioneins; this expression had diminished by the time we analysed rats euthanised on day 82, when an increase in apoptosis was observed. The first cellular changes identified were activated microglia followed by astrogliosis and overexpression of GFAP and S100B proteins.
ConclusionOur data suggest that ALS could spread through CSF and that intracerebroventricular administration of cytotoxic ALS-CSF provokes changes similar to those found in sporadic forms of the disease.
La exposición de líquido cefalorraquídeo (LCR) de pacientes con esclerosis lateral amiotrófica (ELA) induce efectos citotóxicos en cultivos celulares de neuronas motoras in vitro.
Material y métodosSe seleccionó LCR de 32 pacientes con ELA que previamente habían demostrado efectos citotóxicos. Se implantaron con minibombas osmóticas intracerebroventriculares (ICV) en 28 ratas macho adultas y se dividieron en 3 grupos: 9 ratas de LCR de pacientes no-ELA, 15 ratas de ELA-LCR citotóxico y 4 ratas de una solución salina fisiológica. El LCR se administró por vía ICV de forma continua durante periodos de 20 o 43días. Se realizó la evaluación clínica, electromiográfica y análisis de tejidos después de sacrificio a los 20, 45 y 82días tras la cirugía.
ResultadosLos estudios inmunohistoquímicos muestran daño en los tejidos con características similares a las encontradas en formas esporádicas de ELA, tales como sobreexpresión de cistatinaC, transferrina y la proteína en el TDP-43 citoplasmática. Los primeros cambios observados parecían jugar un papel protector por la sobreexpresión de periferina, panAKT, fosfoAKT y metalotioneínas; esta expresión habría disminuido al momento de analizar las ratas que se sacrificaron al día 82, en el que hay un aumento de apoptosis. Los primeros cambios celulares identificados fueron la constatación de activación de la microglía seguido por astrogliosis con sobreexpresión de GFAP y proteína S100B.
ConclusionesNuestros datos parecen indicar que la ELA podría propagarse a través del LCR, y que la administración ICV de ELA-LCR citotóxico produce cambios similares a los encontrados en las formas esporádicas de la enfermedad.
Amyotrophic lateral sclerosis (ALS) is characterised by a selective loss of motor neurons of the brain, brainstem, and spinal cord.1 ALS is sporadic in the majority of cases, although approximately 10% of patients have a family history of the condition2; 20% have mutations of the copper/zinc superoxide dismutase 1 (SOD1) gene.3 Both sporadic and familial ALS may be associated with alterations of the TDP-43 and FUS/TLS proteins.4,5 The majority of cases are linked to an intronic expansion of the GGGGCC hexanucleotide repeat in the C9ORF72 gene.6
The mechanism underlying the selective vulnerability and death of motor neurons in ALS is yet to be identified. The clinical progression pattern observed in mutant SOD1 transgenic mice may suggest that the disease spreads centrifugally from affected areas to adjacent unaffected regions. We may therefore hypothesise that the toxic products generated cause the lesion to spread, affecting adjacent neurons and propagating the disease; the cerebrospinal fluid (CSF) may serve as a vehicle for these factors.7 This concept gave rise to experiments by various researchers into whether in vitro incubation of cultures of neurons and neuronal cell lines with CSF from patients with ALS (ALS-CSF) causes cell damage. This hypothesis is confirmed by the majority of studies,7–22 with some exceptions.23 Our study group has previously observed that ALS-CSF affects the viability of rat brain cortical motor neurons maintained in primary cultures; however, this effect seems to be mediated by a mechanism other than glutamate, although ALS-CSF did increase basal cytosolic Ca2+ concentration in motor neurons.24,25 These findings support the hypothesis that one or more toxic factors present in ALS-CSF are responsible for the damage caused to motor neurons in in vitro experiments; however, it is possible that this mechanism is not responsible for the damage in all patients or in all stages of the disease.26 The question at issue is whether ALS-CSF may also cause cerebral damage in vivo. Acute intrathecal or intracerebroventricular (ICV) injection of ALS-CSF alters neuronal activity, glutamate receptor and ion channel expression, and protein phosphorylation only 2 days after injection27–29; it may also cause mitochondrial alterations, oxidative stress, and lysosomal dysfunction.30
These experiments with local injections do not imitate the time course of ALS, and therefore do not accurately reproduce the disease. For this reason, we considered it beneficial to examine the histopathological and functional changes to the brain after periods of 20 or 43 days of continuous ALS-CSF infusion (via an osmotic minipump connected to the rat's lateral ventricle by a cannula), with a view to determining the consequences of more prolonged exposure to cytotoxic ALS-CSF (cALS-CSF). Although we found no functional changes similar to those observed in ALS, we did find greater early expression of neuroprotective signalling molecules, an early neuroinflammatory reaction accompanied by microglial activation and astrogliosis, and overexpression of such ALS-linked cytoplasmic proteins as TDP-43, cystatin C, and transferrin.
Materials and methodsCSF collection and the preparation of cytotoxic ALS-CSFLumbar puncture was used to collect CSF samples from 32 patients diagnosed with ALS according to the El Escorial diagnostic criteria.31 Between 1.5 and 3cc of fluid were extracted from each patient; samples were centrifuged at 1500rpm for 10minutes then divided into 4 or 5 aliquots for the purposes of the study. Informed consent was granted by all patients. Sixteen patients were women; mean age at the time of CSF collection was 59 years. Four patients had familial ALS; the remaining cases were sporadic. Onset was bulbar in 11 cases, spinal in 20, and bulbospinal in one. Only 4 patients had SOD1 mutations; one of these was asymptomatic. Therefore, patients with SOD1 mutations represented a small group within the sample. Twenty samples (68.7%) were determined to have high levels of cytotoxicity, using the method described below. The control CSF samples (non-ALS-CSF) were obtained from patients experiencing headache or seizures, who underwent lumbar puncture during a routine examination. Control patients also granted informed consent. CSF samples were stored at −80°C prior to use.
Cytotoxicity was demonstrated through the in vitro motor neuron culture experiments described previously.24 The neurons used in these experiments were obtained from the motor cortex of 20-day-old rat embryos and seeded at a density of 105cells/mL in 48-well plates containing 0.25mL of neurobasal medium with B-27; the cultures were allowed to grow for 8-10 days. After this period, the cultures were incubated for 24hours with 10% ALS-CSF; the viability of the neurons was determined using MTT assays.32 The animals were administered ALS-CSF exhibiting cytotoxicity in vitro (at least 20% of motor neurons lost); the majority of samples in this group came from patients with sporadic ALS. Non-cytotoxic CSF samples from 3 patients were also considered prior to ICV administration.
AnimalsAnimals were manipulated following the ethical standards of the Spanish Ethics Committee (RD 1201/2005) and European Directive 86/609/EEC on the protection of animals used for scientific purposes. The experimental procedure was approved by the Ethics Committee for the treatment and use of experimental animals at Hospital Clínico San Carlos (Madrid). Twenty-eight male Wistar rats were procured from Charles River Laboratories. The animals were individually caged under standard colony conditions at the hospital's vivarium, with free access to food and water and a 12:12 light/darkness cycle. CSF was administered to the rats in 2 periods. In period 1, the subjects were 14 rats aged between 1 and 5 months (mean, 3.25 months); in period 2, 14 rats aged between 2.5 and 5 months (mean, 4.1 months) were used. The interval between periods was approximately 3 months.
Experimental groupsThe study included 3 experimental groups. Group 1 included 9 rats, which were implanted with ICV osmotic minipumps administering non-ALS-CSF. Group 2 included 15 rats, which were implanted with ICV minipumps administering cALS-CSF. Finally, group 3 comprised 4 rats implanted with ICV minipumps administering physiological saline solution (sham group).
Intracerebroventricular administration of cytotoxic ALS-CSF and control CSFWe anaesthetised the rats with tribromoethanol, then subcutaneously implanted an osmotic minipump (pumping rate 0.15μL/hour; Alzet 2006 model, DURECT Corporation, Cupertino, USA) into the interscapular area. Prior to implantation, the osmotic minipumps were filled with the corresponding preparation for each experimental group. The minipump was attached to a brain infusion cannula (Alzet) via a polyethylene tube and immersed in normal saline solution at 37°C for 24hours. These procedures were performed under sterile conditions. The cannula was implanted in the right lateral ventricle (−0.5mm anteroposterior, −1.4mm mediolateral, and −33mm dorsoventral; coordinates based on the Paxinos and Watson rat brain atlas)33 and affixed with dental cement.
The pump's reservoir capacity allows a pumping duration of at least 42 days. CSF was infused intrathecally at a rate of 0.15μL/hour for 43 days, except in the case of the rats euthanised at day 20. The remaining study animals, which were euthanised on day 82, were implanted with a mechanically sealed polyethylene tube to prevent local irritation due to the continuous attraction of water into the pump.
Clinical evaluationThe animals underwent clinical evaluation at least once per week, starting one week after surgery. All tests were performed by blinded examiners. We determined body weight, response in the inclined plane test, and motor activity according to the scale proposed by Matsumoto et al.34 Body weight was measured every 4 weeks on a digital scale. In the inclined plane test, rats were placed horizontally, perpendicular to the major axis of the inclined plane; we recorded the maximum angle at which they were able to stay on the plane for 5seconds without falling. In order to take equal measurements for all limbs, the test was performed with the rats in both left-headed and right-headed positions. Scores below 70° on the inclined plane test are well correlated with the onset of muscle weakness in SOD1 (G93A) transgenic rats.34 The Matsumoto et al.34 motor scale tests animals’ ability to right themselves within 30seconds of being laid on their side (righting reflex). Rats with intact bilateral righting reflex were observed inside their cages for one minute. Rats showing limited movement in their cages were moved to another cage to encourage exploration. Rats displaying normal results in these assessments underwent detailed evaluation to identify any observable functional deficit, such as paralysis of the limbs or symptoms of generalised muscle weakness in the open field test. A score of 5 reflects a normal animal, whereas a score of 0 indicates that the rat is completely paralysed. The motor scale has been demonstrated to be correlated with the loss of spinal neurons in a mutant SOD1 (G93A) transgenic mouse model of ALS.34
Electromyography studyOn day 82, both the cALS-CSF and the non-ALS-CSF groups of rats underwent an electromyography (EMG) study, which was performed by a neurologist specialising in the technique. The needle was inserted into the front and rear limbs and into the paravertebral muscles to detect signs of denervation in the form of spontaneous insertion activity (positive sharp waves, fibrillations, or fasciculation). During this procedure, the animals were sedated with 1.5% isoflurane in 0.7L/min oxygen.
EuthanasiaOn days 20, 45, or 82 after surgery, the rats in each group underwent deep anaesthesia (pentobarbital at 60mg/kg and fentanyl at 0.3mg/kg). We injected 10μL of Evans blue dye through the ventricular catheter in order to verify correct placement. The animals were euthanised by intracardiac perfusion with 0.9% saline solution followed by 4% paraformaldehyde in 0.1M phosphate buffer solution. Following the perfusion, the brain and spinal cord were removed, washed with 0.1M phosphate buffer solution, and cryoprotected by immersion in 30% sucrose and optimum cutting temperature solution. The tissue was stored at −80°C prior to use. We cut 50μm cryosections of the motor cortex and the cervical and lumbar segments of the spinal cord (C5-C6 and L3-L5). The sections were placed in a cryoprotectant solution for nervous tissues, containing ethylene glycol and dimethyl sulphide. Of the 9 rats in the non-ALS-CSF group, one was euthanised on day 20, 7 on day 45, and one on day 82. Of the 15 rats in the cALS-CSF group, 4 were euthanised on day 20, 7 on day 45, and 6 on day 82. Finally, of the 4 rats in the sham group, one was euthanised on day 20, 2 on day 45, and one on day 82.
Histochemical and immunofluorescence analysisSections were washed with phosphate buffer solution, permeabilised with 0.1% Triton X-100, and blocked with 10% normal goat serum. The following antibodies were applied overnight at a temperature of 4°C: anti-peripherin (1:200, Millipore, AB 9282), anti-S100B (1:200, Millipore, 04-1054), anti-caspase-3 (1:200, Millipore, 04-1090), anti-glial fibrillary acidic protein (GFAP) (1:500, DakoCytomation, Z0334), anti-GTL1 (1:200, Millipore, AB 1783), anti-GFAP (1:600, Millipore, MAB360), anti-TARDBP (1:200, Abcam, ab42474), anti-ubiquitin (1:100, Abcam, ab7780), anti-transferrin receptor (1:200, Abcam, ab22391), anti-choline acetyltransferase (1:100, Abcam, ab68779), anti-metallothionein (1:100, Abcam, ab12228), anti-stefin (1:100, Abcam, ab68290), anti-MHC-II (1:500, Abcam, ab6403), anti-Iba1 (1:1000, Wako, 019-19741), anti-pan-Akt (Cell Signalling Technology, 2920S), and anti-phospho-Akt (Cell Signalling Technology, 4060S). The slides were washed 3 times in phosphate buffer solution. Primary antibodies were tested with Cy3 (1:1000, Jackson) or with Alexa Fluor secondary antibody conjugates 488, 555, or 647 (1:500, Invitrogen). The sections were washed 3 times in phosphate buffer solution, contrasted with DAPI, and mounted with Fluorsave reagent (Calbiochem).
Quantification of immunohistochemical dataAn Olympus FV1000 confocal microscope was used to obtain fluorescence images. A descriptive histochemical study was performed for the 3 established periods, analysing the changes observed over time and between groups (CSF groups vs control group) for the immunohistochemical markers observed. Histological analysis assessed the areas associated with ALS, specifically the motor cortex, medulla oblongata, and the C5-C6 and L3-L5 spinal cord segments.
GFAP, Iba1, caspase-3, and MHC-IIAll confocal images were acquired with the same fluorescence distribution and configuration, using the Olympus confocal software (Olympus FluoView FV1000). Quantitative analysis was performed using the ImageJ software (version 1.42q; NIH, USA). We counted the number of positively stained cells, randomly selecting a 368μm2 region in each area analysed from 3 or 4 coronal sections.
Peripherin, S100B, pan-Akt, phospho-Akt, and metallothioneinPeripherin, S100B, pan-Akt, phospho-Akt, and metallothionein were measured according to a modified version of the physical disector method described by Reed and Howard,35 using the principle of the unbiased sampling brick of the 3D disector. The analysis was performed using optical density measurements (binary black and white image processing), resulting in a percentage value for the field analysed. A blinded researcher randomly selected 8 areas per animal using the ImageJ software.
Statistical analysisData were processed using the GraphPad Prism 5 application and are reported as mean±standard error (SE). Graphs were created using the same program. Statistical significance was set at P<.05.
ResultsAll animals survived ICV osmotic minipump implantation; none showed any sign of infection (abscess, oedema, pus in the area of the implant); no animal displayed seizures, aggression, piloerection, or postration. The infusion was performed for a period of either 20 or 43 days, and the time course of the effects of CSF or saline infusion was assessed at days 20, 45, and 82 after implantation of the osmotic minipump.
Clinical and electromyographic examinationNo significant differences were observed between cALS-CSF rats and the other groups for any clinical variable, including body weight, behaviour during the inclined plane test, and Matsumoto motor score; normal results were observed for all variables. In the EMG study, we observed positive sharp waves in the front and rear limbs and in the paravertebral muscles, without fibrillation or fasciculation potentials, in 6 rats from the cALS-CSF group at 82 days after implantation; no denervation pattern was observed, however. Control rats showed normal EMG results.
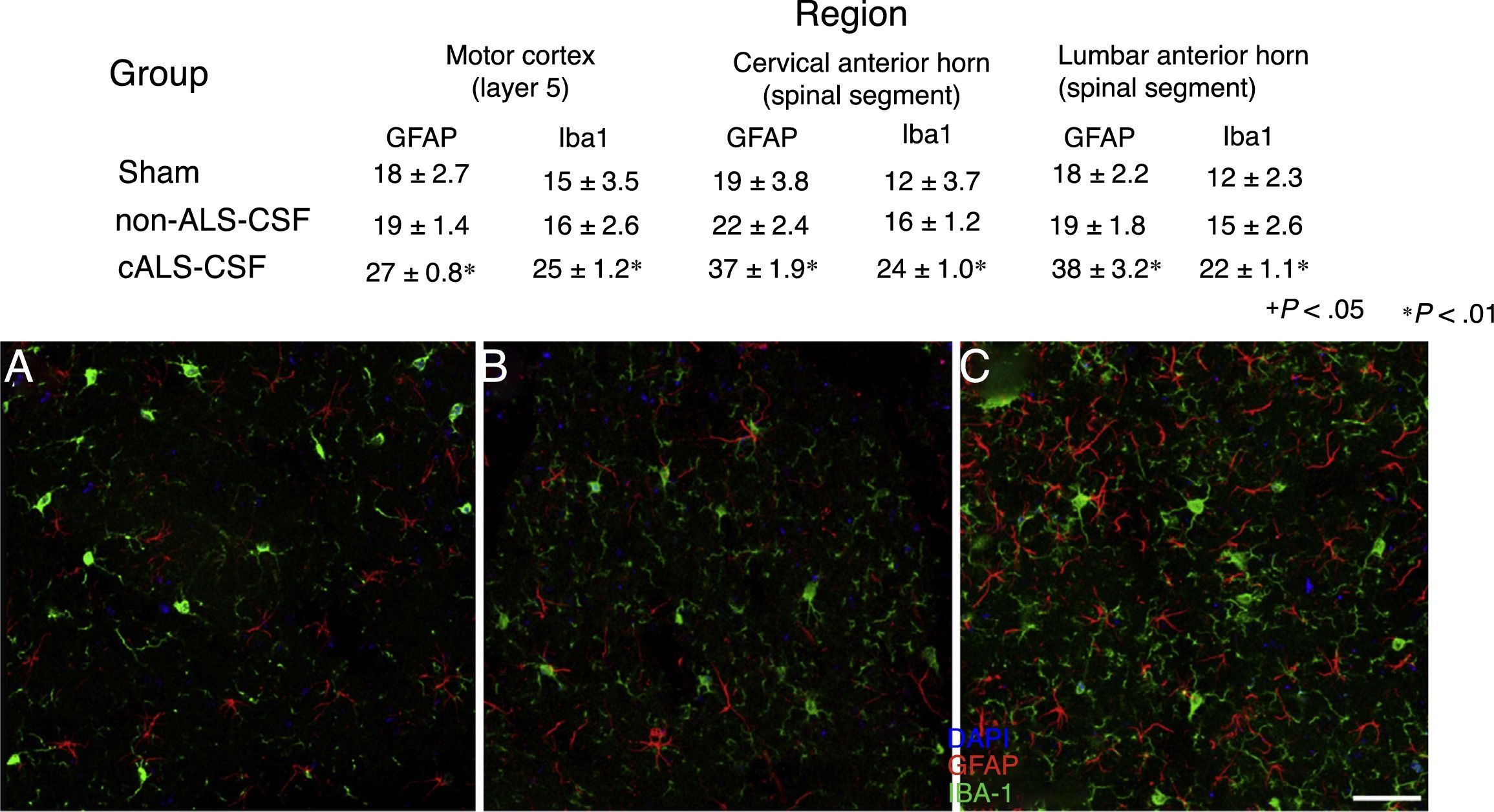
Microglial activation and GFAP, S100B, GTL1, and caspase-3 expressionReactive gliosis, microglial activation, and caspase-3 expression were observed in the motor cortex in all 3 experimental groups on day 20 after implantation. These findings may be due to an inflammatory response secondary to the surgical implantation of the pumps. At day 45, the cALS-CSF group showed a considerable increase in glial reactivity and in microglial response in all regions (Figs. 1 and 2). In the spinal cord segments of cALS-CSF rats, some motor neurons were surrounded by microglia. Immunohistochemical quantification of the Iba1 marker revealed alterations to microglial morphology; the cells passed from a resting state characterised by microglial somata displaying thin, ramified processes, to activated microglia with few ramifications. Cell bodies were longer, with longer, thicker processes than at post-implantation day 20. On days 45 and 82, we observed amoeboid microglia, with round bodies and short, thick, robust processes, suggesting activation of phagocytic cells in the vicinity of the motor neurons. These cells expressed MHC-II, a marker of inflammation. MHC-II expression was 3 times lower in the sham and non-ALS-CSF groups than in the cALS-CSF group (Fig. 3). One interesting observation was the pronounced expression of astrocytes and the presence of hypertrophic astrocytes displaying GFAP overexpression in the vicinity of cortical and spinal motor neurons. The changes observed were more pronounced at day 45. MHC-II expression was also observed at day 82, but at much lower levels (Fig. 3). Unlike the cALS-CSF group, the other 2 groups displayed reduced GFAP and caspase-3 expression, even at day 82 after implantation. A minor increase was also observed in the glutamate transporter GTL1, which has been associated with ALS; ALS-CSF modifies the expression of this protein.36 In the cALS-CSF group, GTL1 was detected near the spinal motor neurons; this was correlated with GFAP expression, a marker of astrocyte activation. S100B overexpression was observed in the cALS-CSF group only on days 45 and 82.
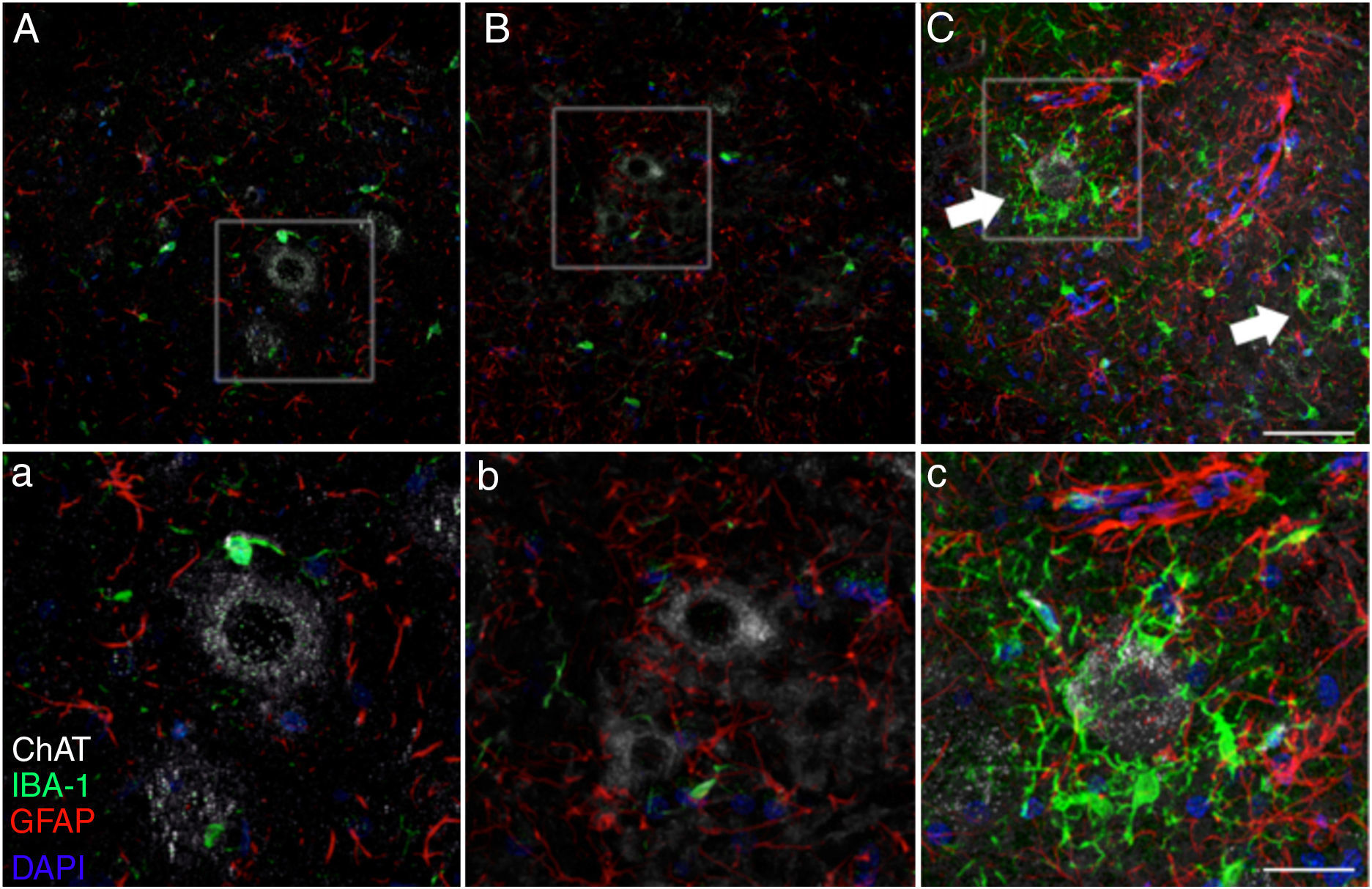
 of rats in the cALS-CSF group displayed greater expression of GFAP, with numerous hypertrophic astrocytes (panels C, c). Furthermore, we identified a higher density of Iba1+ cells with enlarged processes; these were often in close proximity to or even surrounding the neurons (arrows in panel C; detail in panel c). These changes were not observed in the sham or non-ALS-CSF groups, in which isolated microglial cells were occasionally observed close to motor neurons. Scale bar: A–C, 60 μm; a–c, 30 μm.")
Forty-five days after surgery, the increased glial reactivity and microglial response in the cALS-CSF group was evident in all regions studied. For example, the anterior horn (lumbar segment) of rats in the cALS-CSF group displayed greater expression of GFAP, with numerous hypertrophic astrocytes (panels C, c). Furthermore, we identified a higher density of Iba1+ cells with enlarged processes; these were often in close proximity to or even surrounding the neurons (arrows in panel C; detail in panel c). These changes were not observed in the sham or non-ALS-CSF groups, in which isolated microglial cells were occasionally observed close to motor neurons. Scale bar: A–C, 60 μm; a–c, 30 μm.
. There is a visible increase in the number of Iba1+ and GFAP+ cells in animals from the cALS-CSF group (C) compared to the sham (A) and non-ALS-CSF (B) groups. Motor cortex layers 4-6, 45 days after surgery. Scale bar: 50μm. Values expressed as mean±SD.")
Confocal micrographs of the motor cortex, demonstrating the data presented in the tables (from different study periods). There is a visible increase in the number of Iba1+ and GFAP+ cells in animals from the cALS-CSF group (C) compared to the sham (A) and non-ALS-CSF (B) groups. Motor cortex layers 4-6, 45 days after surgery. Scale bar: 50μm. Values expressed as mean±SD.
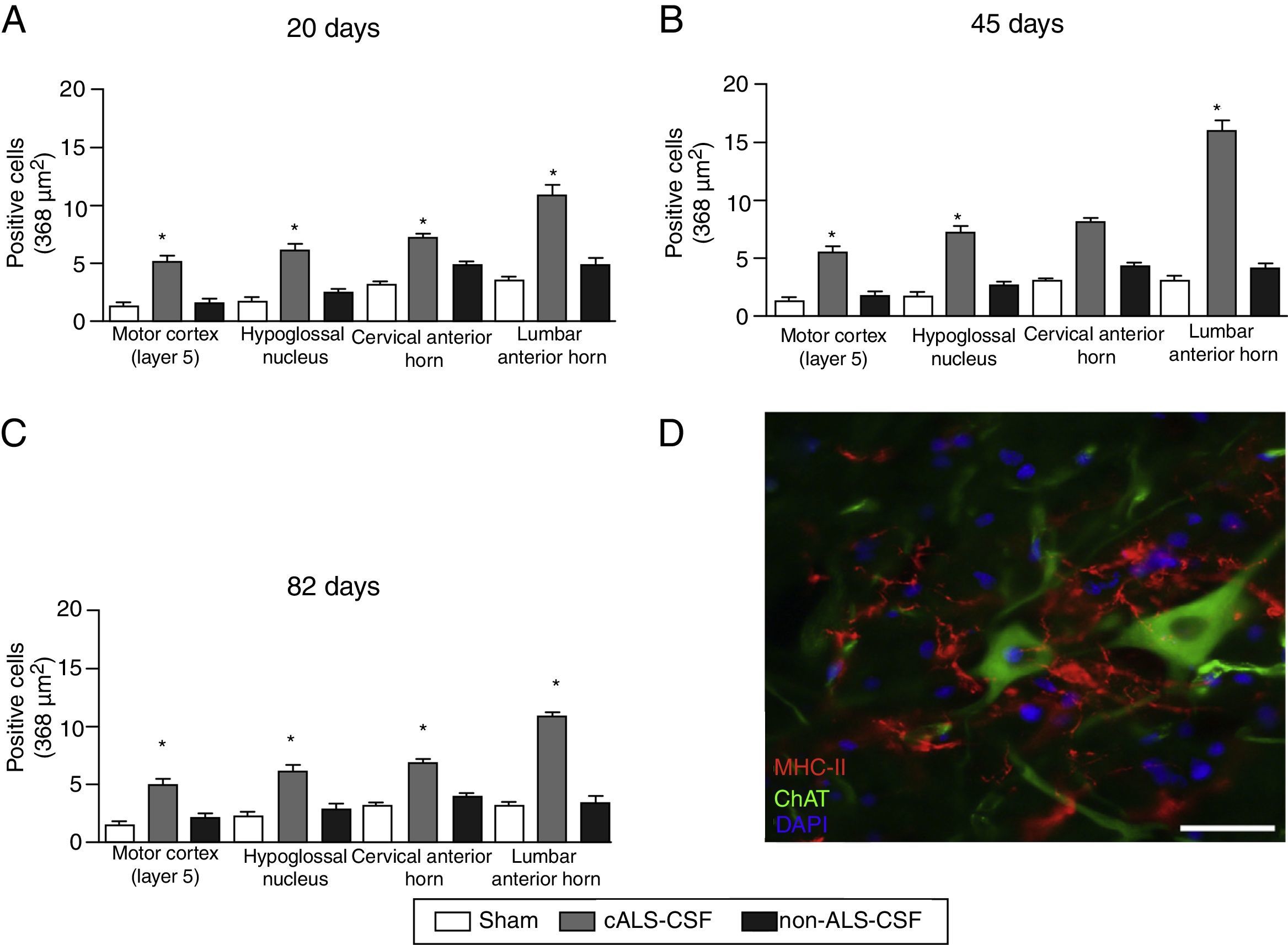
 Quantitative analysis of MHC-II expression in different regions of the brain and spinal cord in the 3 time periods studied. The cALS-CSF group displayed a significant increase in the numbers of microglial cells, which is clear evidence of a neuroinflammatory process. (D) MHC-II+ cells in the lumbar spinal cord. These cells were also observed in the vicinity of the motor neurons. Scale bar: 50 μm. * P<.05.")
(A–C) Quantitative analysis of MHC-II expression in different regions of the brain and spinal cord in the 3 time periods studied. The cALS-CSF group displayed a significant increase in the numbers of microglial cells, which is clear evidence of a neuroinflammatory process. (D) MHC-II+ cells in the lumbar spinal cord. These cells were also observed in the vicinity of the motor neurons. Scale bar: 50 μm. * P<.05.
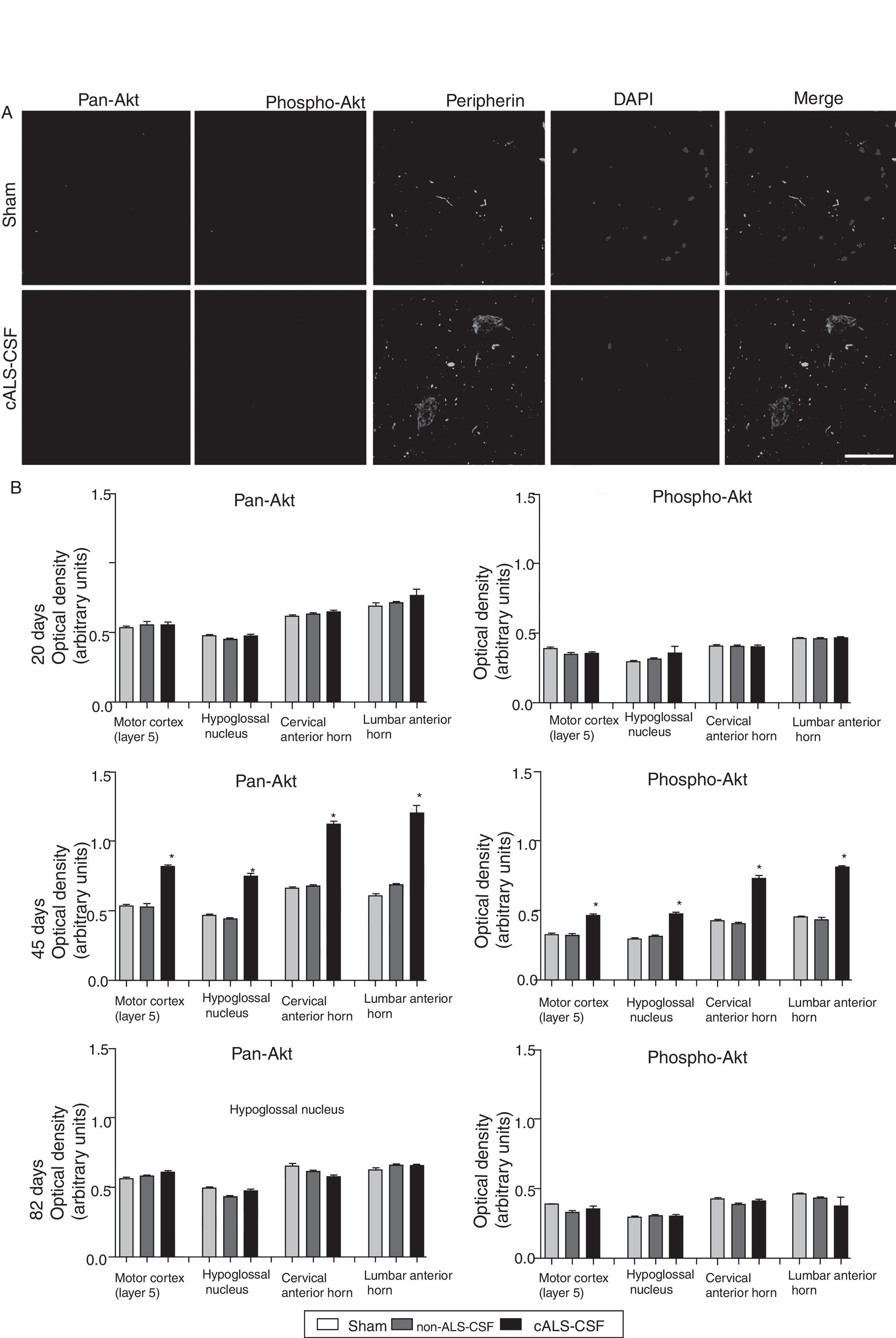
Pan-Akt and phospho-Akt expression were quantified in order to assess whether the increased caspase-3 expression was associated with increased expression of these proteins, which are strongly linked to cell survival and resistance to apoptosis. The sham and non-ALS-CSF groups showed no significant change in the expression of these proteins at any point over the course of the study. The cALS-CSF group displayed significant increases in pan-Akt and phospho-Akt expression at day 45 (expressed at levels 3 times greater than those observed in the other groups); however, the levels observed at day 82 were similar to those for the sham group (Fig. 4).
 Confocal micrographs displaying pan-Akt, phospho-Akt, and peripherin expression. (B) Optical density quantification of phospho-Akt and pan-Akt. Increased pan-Akt and phospho-Akt expression were observed at day 45 in the cALS-CSF group (A and B). An interesting observation in the cALS-CSF group was the increased neuronal expression of peripherin, phospho-Akt, and pan-Akt (A). On post-surgery day 82, pan-Akt and phospho-Akt concentrations had decreased to similar levels to those observed in the sham and non-ALS-CSF groups (B). Scale bar: 50 μm.")
Pan-Akt and phospho-Akt expression in various regions of the central nervous system in the 3 study groups. (A) Confocal micrographs displaying pan-Akt, phospho-Akt, and peripherin expression. (B) Optical density quantification of phospho-Akt and pan-Akt. Increased pan-Akt and phospho-Akt expression were observed at day 45 in the cALS-CSF group (A and B). An interesting observation in the cALS-CSF group was the increased neuronal expression of peripherin, phospho-Akt, and pan-Akt (A). On post-surgery day 82, pan-Akt and phospho-Akt concentrations had decreased to similar levels to those observed in the sham and non-ALS-CSF groups (B). Scale bar: 50 μm.
Peripherin plays an important role in axonal transport in spinal motor neurons. It has recently also been associated with certain typical neuropathies of sporadic ALS due to its presence in Bunina bodies.37 We therefore considered it relevant to assess peripherin expression in motor neurons. All the sections from the cALS-CSF group displayed motor neurons positive for peripherin expression (Figs. 5 and 6), although levels of the protein were 4 times higher in the animals euthanised at day 45 than in the other study groups and periods (Fig. 6). Cells with greater peripherin immunoreactivity also co-expressed pan-Akt and phospho-Akt. These cells displayed peripherin in a filament pattern; this is also observed in normal conditions. The cALS-CSF group also displayed the marker in cytoplasmic inclusions reminiscent of coarse or granular structures, which may trigger the formation of precipitates or inclusions of proteins (Fig. 5).
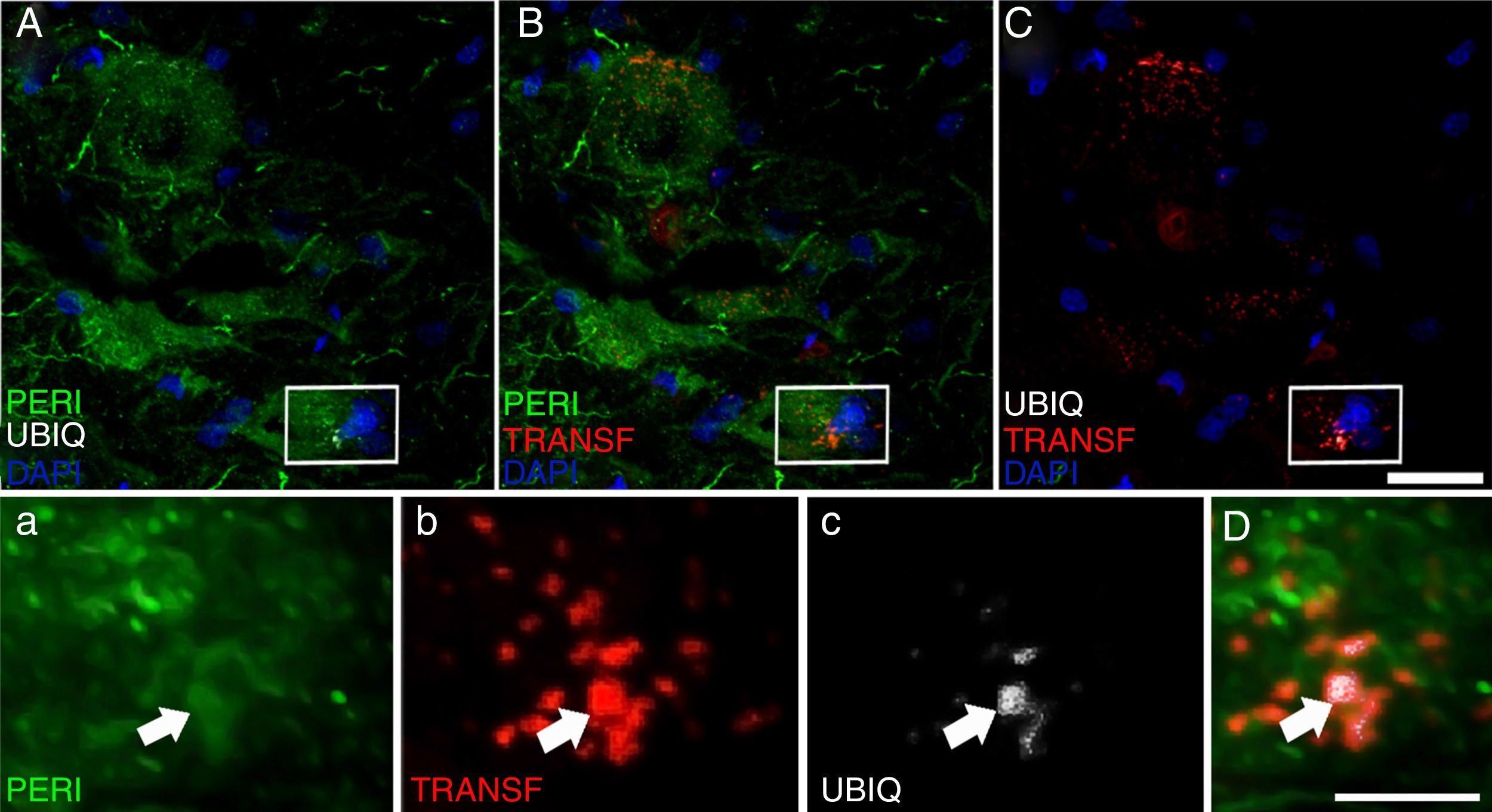
, ubiquitin (UBIQ), and transferrin (TRANSF) expression following cALS-CSF infusion. On post-surgery days 45 and 82, animals treated with cALS-CSF displayed cytoplasmic inclusions and ubiquitin-positive neurons. Ubiquitin inclusions (arrows) co-localised with peripherin, and occasionally with transferrin. Scale bar: A-C, 20μm; a-c and D, 2μm.")
Peripherin (PERI), ubiquitin (UBIQ), and transferrin (TRANSF) expression following cALS-CSF infusion. On post-surgery days 45 and 82, animals treated with cALS-CSF displayed cytoplasmic inclusions and ubiquitin-positive neurons. Ubiquitin inclusions (arrows) co-localised with peripherin, and occasionally with transferrin. Scale bar: A-C, 20μm; a-c and D, 2μm.
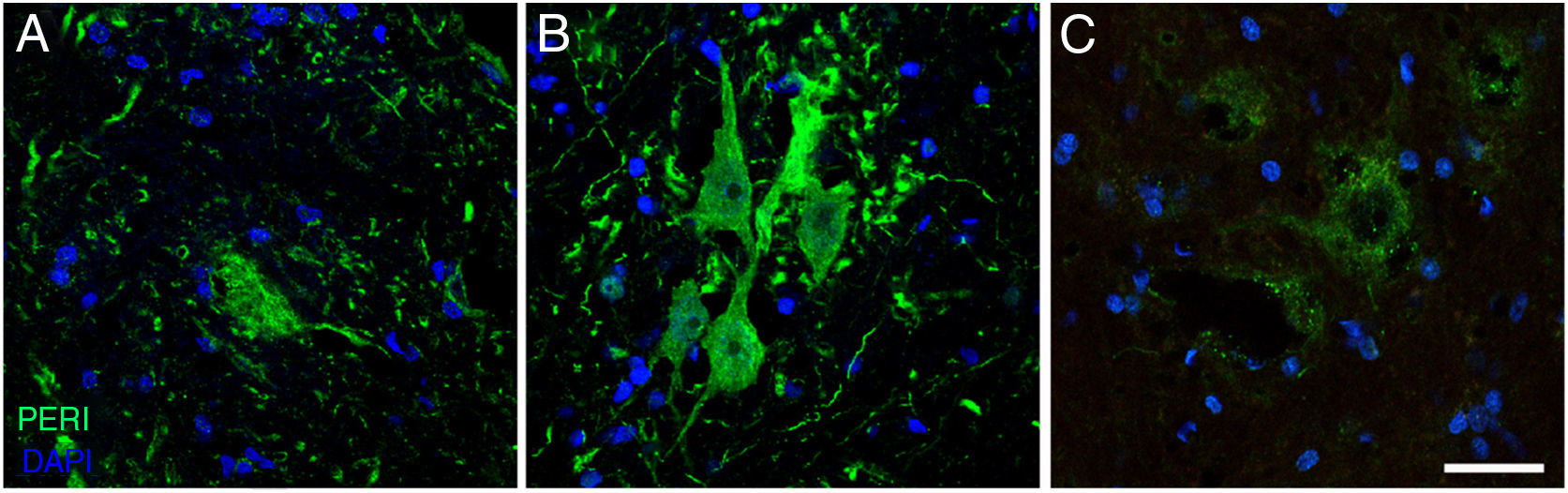
 expression in the cALS-CSF group over the course of the study (A: 20 days; B: 45 days; C: 82 days). A temporary increase in peripherin expression was observed in motor neurons at day 45; this returned to baseline conditions at day 82. Increased peripherin expression has been associated with spinal cord lesions; under normal circumstances, the protein is expressed at lower levels. Scale bar: 40 μm.")
Immunofluorescence images showing peripherin (PERI) expression in the cALS-CSF group over the course of the study (A: 20 days; B: 45 days; C: 82 days). A temporary increase in peripherin expression was observed in motor neurons at day 45; this returned to baseline conditions at day 82. Increased peripherin expression has been associated with spinal cord lesions; under normal circumstances, the protein is expressed at lower levels. Scale bar: 40 μm.
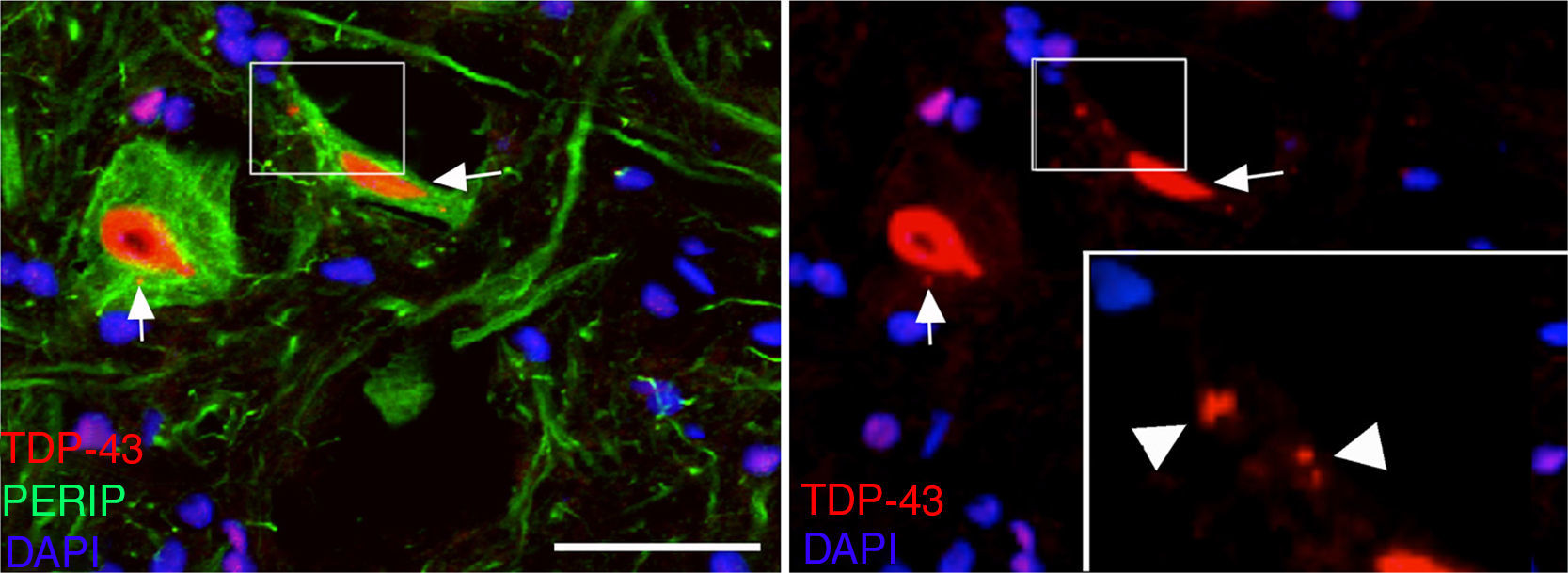
At post-surgery days 45 and 82, the cALS-CSF group displayed cells with increased ubiquitin, transferrin (Fig. 5), and cystatin C expression. These findings are closely associated with sporadic forms of ALS.38 We also observed translocation of TDP-43 in the cytoplasm of motor neurons (Fig. 7). Interestingly, immunohistochemical analysis revealed inclusions of TDP-43, ubiquitin, and cystatin C; these proteins were co-localised in motor neuron cytoplasm. At day 82, there was increased presence of these inclusions and reduced peripherin expression (Fig. 6).
Metallothioneins in the spinal cord neurons of animals receiving cALS-CSF. These inclusions can be observed in the inset detailed image (arrowheads). Scale bar: 40μm.")
As some studies have suggested that alterations in the homeostasis of copper and zinc may play a role in ALS pathogenesis, we studied the expression of metallothioneins.39,40 Metallothionein expression was related to the time course of changes following surgery (days 45 and 82), and was observed only in the cALS-CSF group, in motor neurons displaying co-expression of ubiquitin and transferrin. The increased S100B expression at days 45 and 82 was strongly related with increased peripherin expression near motor neurons. S100B expression was observed in GFAP+ astrocytes of neutrophils adjacent to motor neurons. S100B-expressing astrocytes are strongly related to the balance of calcium, copper, and zinc. S100B is a calcium-binding protein expressed exclusively in glial cells.
DiscussionThis study aimed to determine the effects of prolonged ALS-CSF administration to rats in order to gain insight into the effects that may occur in patients with CSF cytotoxicity. This demonstrates that prolonged ICV infusion of ALS-CSF in rats causes similar cytohistochemical changes in the brain and spinal cord to those observed in patients with sporadic ALS. These changes were not observed in rats infused with saline solution or with non-ALS-CSF. Infusion was maintained for either 20 or 43 days and tissues were analysed 20, 45, or 82 days after implantation of the cannula. The animals that were euthanised on day 82 received the infusion only for the first 43 days post-implantation.
The significant changes observed after 43 days of infusion were microglial activation followed by astrogliosis. Microglial activation and proliferation prior to the onset of clinical signs is also observed in patients with ALS41 and in SOD1 (G93A) transgenic mice.42 Furthermore, activated microglia appear earlier than neuronal loss in rat models of ALS.43 This is consistent with the observation that reduced SOD1 (G93A) expression in microglial cells slows disease progression and prolongs survival.44 Microglial cells expressing SOD1 (G93A) show increased neurotoxicity and increased production of NO and the cytokine MCP-1 following in vitro activation of the microglia.45 These findings support the hypothesis that microglial activation plays an important role in ALS pathogenesis.46,47 Microglial cells were observed in close contact with spinal neurons 45 days after minipump implantation (43 days of ALS-CSF infusion). These cells displayed the typical morphology of activation, in addition to increased expression of MHC-II, which is believed to be a marker of complement-dependent inflammatory response.48,49 Astrogliosis is also evident, although the nature of its involvement in ALS pathogenesis and whether it appears before or after microglial activation remains unknown. Another noteworthy finding was astrocytic S100B overexpression at 45 and 82 days. This protein has been associated with survival in patients with ALS50,51 and is regulated by spinal motor neurons and astrocytes both in patients52 and in rats exposed to ALS-CSF for 48hours.53 Astrocytes with inclusions have also been observed in mutant SOD1 mice.54 We also observed astrogliosis and GFAP overexpression following microglial activation. This may support the idea of a neuroinflammatory mechanism as an early event in ALS pathogenesis.
In recent years, emphasis has been placed on the cytoplasmic translocation of TDP-43, a protein encoded by the TARDBP gene; mutations in this gene have been associated with ALS.4 We observed TDP-43 in the ubiquitin inclusions in rats treated with CSF from patients with sporadic ALS, but not those treated with CSF from patients with SOD1 mutations. We observed cytosolic inclusions of TDP-43 co-localised with ubiquitin on days 45 and 82 in the cALS-CSF group, which supports the hypothesis that these changes are related to sporadic ALS. This is further supported by the fact that the first changes in motor neurons included overexpression of cystatin C, transferrin, and peripherin, 3 proteins that are closely related to Bunina bodies in sporadic ALS.37,55,56 Peripherin mutations have previously been associated with sporadic ALS.57
Another variable addressed was the PI3K/Akt signalling pathway, which is associated with cell survival.58 Increased pan-Akt and phospho-Akt expression was observed on day 45 in rats receiving cALS-CSF. This finding is interesting given that peripherin is a substrate of PI3K. It should be noted that increased expression of PI3K, but not of Akt, has been detected in the spinal cords of patients with ALS.59 However, increased phospho-Akt expression has been observed in the spinal cords of SOD1 (G93A) transgenic mice in the asymptomatic phase of the disease.60 In vitro studies have also demonstrated PI3K/Akt alterations in cells from mutant SOD1 (G93A) rats.61 Therefore, peripherin and PI3K/Akt may increase in response to cellular stress caused by cALS-CSF. The increased caspase-3 levels (indicative of apoptosis) observed in these rats are compatible with this hypothesis and with the detection of apoptotic motor neurons in patients with ALS.62–64
The last noteworthy finding is the increased metallothionein expression in rats receiving cALS-CSF. This is consistent with reports of higher levels of these proteins in mutant SOD1 mice65–67; a similar increase in metallothionein mRNAs has also been reported in these mice.68 This is consistent with the observation that offspring from crossing ALS mice and metallothionein-deficient mice show accelerated disease progression68; an improvement has been observed in mice with greater metallothionein expression.69 In contrast, the expression of metallothioneins and their mRNAs is lower in patients with ALS.70 Metallothioneins are copper-/zinc-binding proteins that reduce levels of reactive oxygen species and counteract metal toxicity. In this context, greater metallothionein expression may lead to a neuroprotective reaction in response to prolonged cALS-CSF infusion in rats. This is consistent with the increased metallothionein expression found in the spinal motor neurons of ALS model mice, with the objective of avoiding neuronal loss and inhibiting disease progression.71
The histopathological findings we describe are compatible with the hypothesis that CSF cytotoxicity in rats, induced by prolonged ICV infusion of cALS-CSF, causes similar brain and spinal cord changes to those observed in sporadic forms of ALS. Our study has certain limitations. Firstly, clinical symptoms were not observed in any of the 3 study periods (20, 45, or 82 days). In SOD1 (G93A) transgenic ALS mice, the initial clinical symptoms appear at 90-100 days of age72; however, animals with other mutations may begin showing symptoms later.38 The early anomalies observed include Golgi fragmentation at 31 days of age73; subtle changes may also take place at younger ages.74 The first alterations observed in mutant SOD1 mice affect the cervical spinal cord.38 Other changes observed in the cervical and lumbar spinal cord are similar to those affecting the motor cortex. This suggests that the harm caused by cALS-CSF infusion into the lateral ventricle had propagated distally to the spinal cord. This observation is compatible with the hypothesis that ALS progression is mediated in part by the CSF. Secondly, various markers of proteins that were overexpressed at 45 days continued to be detectable at 82 days (peripherin, GFAP, pan-Akt, and phospho-Akt), although at lower concentrations. Others were expressed equally on days 45 and 82 (cystatin C, transferrin, ubiquitin, and TDP-43 translocation). However, the differing sample sizes of the study groups and the low number of control rats constitute a limitation. Despite this, abnormal expression of these proteins in the cALS-CSF group was evident; the different results observed in the control group confirm that the changes seen are not artefacts of the method. The ICV infusion of cALS-CSF lasted 43 days, the maximum period allowed by the volume of the osmotic minipump. Despite the fact that cALS-CSF was not administered between days 43 and 82, apoptotic motor neurons were only observed in animals examined on day 82. This suggests that the pathological process may remain active beyond day 43, for which reason the extension of cALS-CSF infusion beyond day 43, and/or following the time course of the disease for longer than 82 days, may reveal clinical motor manifestations of the disease similar to those observed in sporadic ALS.
In conclusion, this study presents the early effects of ALS-CSF cytotoxicity following weeks of prolonged ICV infusion of ALS-CSF confirmed to be cytotoxic in a preliminary culture of motor neurons. At days 45 and 82 after infusion was started, the motor cortex, thalamus, and cervical/lumbar spinal cord displayed changes in the expression of various typical markers of ALS in humans. These findings may improve our understanding of ALS pathogenesis and progression and assist in identifying new therapeutic targets for the development of new compounds that could slow or even halt the progression of the disease.
FundingThis study was funded by 4 grants awarded to JMG by Mutua Madrileña, Spain, in 2008 and 2009. Further funding was awarded to AGG by the following Spanish institutions: (1) NDE07/09, Agencia Laín Entralgo, Region of Madrid; (2) Fundación Eugenio Rodríguez Pascual; (3) Fundación CIEN, Instituto de Salud Carlos III; (4) RENEVAS-RETICS-RD06-026, Instituto Carlos III; (5) SAF 2010-21795, Spanish Ministry of Economy, Industry, and Competitiveness; and (6) MAT2011-28791-C03.
Conflicts of interestThe authors have no conflicts of interest to declare.
The authors wish to thank Pablo González and María Cruz Rodríguez for coordinating veterinary procedures, as well as the Fundación Teófilo Hernando, for their constant support.
Please cite this article as: Gómez-Pinedo U, Galán L, Yañez M, Matias-Guiu J, Valencia C, Guerrero-Sola A, et al. La infusión intracerebroventricular prolongada de líquido cefalorraquídeo procedente de pacientes con esclerosis lateral amiotrófica provoca cambios histológicos en el cerebro y la médula espinal de la rata similares a los hallados en la enfermedad. Neurología. 2018;33:211–223.
