









A ganglioglioma is a type of primary central nervous system low grade tumour composed of mixed populations of glial and neuroepithelial elements. They account for 0.4–2% of all intracranial tumours and appear more commonly in children and young adults. Seizures, which are the most important symptom in these tumours, improve significantly after surgical excision.
MethodsBetween 1995 and 2008, 20 patients with (12 adults and 8 children) with intracranial ganglioglioma were treated at our hospital. Clinical information obtained by chart review included sex, age at onset of symptoms, clinical history, results of neurological examination, tumour location, CT and MRI appearance, surgical results and follow-up. All patients underwent tumour resection and the extent of surgery was determined from the surgical reports and postoperative imaging studies.
ResultsThe median age of patients was 26.4 years (range, 1–75 years), and the female to male ratio was 1.5:1. Except in one case, all patients had seizures with a median duration before diagnosis of 7.4 years (range 1–29). Seventeen tumours were located in the temporal lobe (9 right and 8 left). Macroscopically complete excision was performed in 17 patients and subtotal in the remaining 3. There were 4 cases of recurrence treated by surgery and radiotherapy being added in one case. The mean follow-up was 8.5 years (range 22 months–14 years) and disease free survival at 5 years was 85% and an overall survival of 95%.
ConclusionsThe seizures, which are the most frequent symptoms, significantly improved after surgical removal. Surgery is the first choice of therapy in these tumours, and in the presence of subtotal resection or tumour recurrence the best indication for treatment is repeat surgery. Radiotherapy should be reserved only for malignant forms.
El ganglioglioma es un tumor primario, de bajo grado, del sistema nervioso central constituido por una población celular mixta de elementos gliales y neuronales. Representan entre el 0.4 al 2% de todos los tumores intracraneales y afectan fundamentalmente a niños y adultos jóvenes.
MétodosEntre los años 1995 y 2008 hemos tratado en nuestro hospital 20 pacientes (12 adultos y 8 niños) con ganglioglioma intracraneal. Revisamos retrospectivamente el sexo, el síntoma de inicio y la edad, sintomatología y tiempo de evolución, exploración neurológica, localización del tumor, aspecto en la tomografía computarizada y resonancia magnética, el tratamiento quirúrgico y la evolución. Todos los pacientes fueron intervenidos quirúrgicamente y la extensión de la resección fue evaluada de la hoja operatoria y del seguimiento neuroradiológico.
ResultadosLa media de edad de los pacientes fue de 26.4 años (rango 1–75) y el ratio mujer varón fue de 1.5:1. Excepto en un caso, todos los paciente debutaron con crisis epilépticas con una duración media antes del diagnóstico de 7. 4 años (rango 1–29). Diecisiete tumores estaban localizados en el lóbulo temporal (9 derechos y 8 izquierdos). Se realizó extirpación macroscópicamente completa en 17 pacientes y subtotal en los 3 restantes. Se presentaron 4 recidivas que fueron tratadas mediante reintervención, añadiendose radioterapia en uno de los casos. El tiempo medio de seguimiento fue de 8.5 años (rango 22 meses–14 años) y la supervivencia libre de enfermedad a los 5 años fue del 85% y la supervivencia global del 95%.
ConclusionesLas crisis epilépticas, que constituyen el síntoma más frecuente mejoran de forma significativa tras la extirpación quirúrgica. El tratamiento quirúrgico es la primera opción terapéutica en este tipo de tumores, y ante la presencia de resecciones subtotales o recidivas tumorales la mejor indicación de tratamiento es la reintervención. La radioterapia debe reservarse únicamente para las formas malignas.
Neoplasms of the central nervous system (CNS) are a very heterogeneous group of tumours with different histologies, behaviours and locations: as such, they require different and complex treatment approaches. In most cases, they are glial tumours of both high grade and low grade or meningeal tumours. The World Health Organization's (WHO) classification includes a group of tumours denominated glioneural or neuroglial tumours, accounting for between 0.4% and 2% of all CNS tumours, and contemplates tumours characterized by the presence of ganglionar cells and glial (astrocytic or oligodendroglial) elements that tend to correspond with WHO grades I and II.1
These tumours characteristically occur in children and young adults; they have a preference for the temporal lobe, and in many cases, course with long-standing epileptic seizures.2,3
We present our department's experience with 20 cases of gangliogliomas (GG) in surgically treated cerebral hemispheres over a 14-year period.
Patients and methodWe conducted a descriptive, retrospective study of 20 patients with supratentorial ganglioglioma, diagnosed and surgically treated at our Neurosurgery Department between 1995 and 2008; 11 cases were adult patients and 9 were paediatric patients (under 16 years of age).
In addition to the basic epidemiological data, we analyzed the first symptom, time of evolution from the onset of symptoms until definitive diagnosis, the most relevant data from the clinical examination, neurophysiological and neuroradiological diagnostic tests performed, the treatment applied and its complications, surgical outcomes and evolution.
Clinical and radiological follow-ups were possible in all patients.
All of the resected specimens were analyzed histologically by means of haematoxylin–eosin, using specific immunohistochemical stains.
ResultsThe 20 tumours analyzed comprise 1.8% of all the neoplastic processes surgically treated at our department over the period of 14 years.
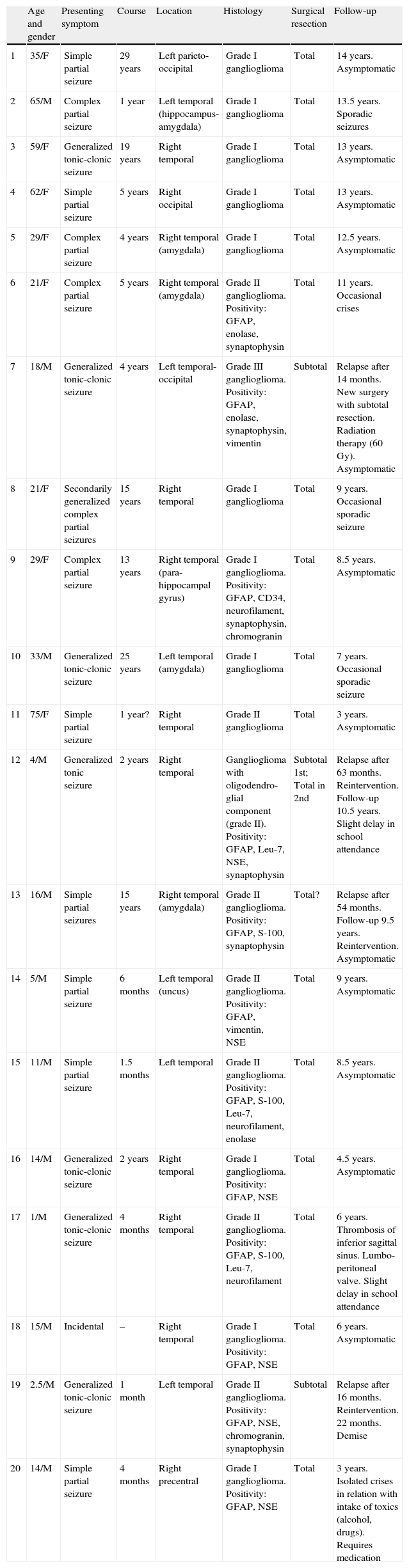
Age and genderThe series consists of 12 males and 8 females, with ages ranging from 1 to 75 years (mean 26.4) (Table 1).
Clinical and neurophysiological data from the series.
| Age and gender | Presenting symptom | Course | Location | Histology | Surgical resection | Follow-up | |
| 1 | 35/F | Simple partial seizure | 29 years | Left parieto-occipital | Grade I ganglioglioma | Total | 14 years. Asymptomatic |
| 2 | 65/M | Complex partial seizure | 1 year | Left temporal (hippocampus-amygdala) | Grade I ganglioglioma | Total | 13.5 years. Sporadic seizures |
| 3 | 59/F | Generalized tonic-clonic seizure | 19 years | Right temporal | Grade I ganglioglioma | Total | 13 years. Asymptomatic |
| 4 | 62/F | Simple partial seizure | 5 years | Right occipital | Grade I ganglioglioma | Total | 13 years. Asymptomatic |
| 5 | 29/F | Complex partial seizure | 4 years | Right temporal (amygdala) | Grade I ganglioglioma | Total | 12.5 years. Asymptomatic |
| 6 | 21/F | Complex partial seizure | 5 years | Right temporal (amygdala) | Grade II ganglioglioma. Positivity: GFAP, enolase, synaptophysin | Total | 11 years. Occasional crises |
| 7 | 18/M | Generalized tonic-clonic seizure | 4 years | Left temporal-occipital | Grade III ganglioglioma. Positivity: GFAP, enolase, synaptophysin, vimentin | Subtotal | Relapse after 14 months. New surgery with subtotal resection. Radiation therapy (60 Gy). Asymptomatic |
| 8 | 21/F | Secondarily generalized complex partial seizures | 15 years | Right temporal | Grade I ganglioglioma | Total | 9 years. Occasional sporadic seizure |
| 9 | 29/F | Complex partial seizure | 13 years | Right temporal (para-hippocampal gyrus) | Grade I ganglioglioma. Positivity: GFAP, CD34, neurofilament, synaptophysin, chromogranin | Total | 8.5 years. Asymptomatic |
| 10 | 33/M | Generalized tonic-clonic seizure | 25 years | Left temporal (amygdala) | Grade I ganglioglioma | Total | 7 years. Occasional sporadic seizure |
| 11 | 75/F | Simple partial seizure | 1 year? | Right temporal | Grade II ganglioglioma | Total | 3 years. Asymptomatic |
| 12 | 4/M | Generalized tonic seizure | 2 years | Right temporal | Ganglioglioma with oligodendro-glial component (grade II). Positivity: GFAP, Leu-7, NSE, synaptophysin | Subtotal 1st; Total in 2nd | Relapse after 63 months. Reintervention. Follow-up 10.5 years. Slight delay in school attendance |
| 13 | 16/M | Simple partial seizures | 15 years | Right temporal (amygdala) | Grade II ganglioglioma. Positivity: GFAP, S-100, synaptophysin | Total? | Relapse after 54 months. Follow-up 9.5 years. Reintervention. Asymptomatic |
| 14 | 5/M | Simple partial seizure | 6 months | Left temporal (uncus) | Grade II ganglioglioma. Positivity: GFAP, vimentin, NSE | Total | 9 years. Asymptomatic |
| 15 | 11/M | Simple partial seizure | 1.5 months | Left temporal | Grade II ganglioglioma. Positivity: GFAP, S-100, Leu-7, neurofilament, enolase | Total | 8.5 years. Asymptomatic |
| 16 | 14/M | Generalized tonic-clonic seizure | 2 years | Right temporal | Grade I ganglioglioma. Positivity: GFAP, NSE | Total | 4.5 years. Asymptomatic |
| 17 | 1/M | Generalized tonic-clonic seizure | 4 months | Right temporal | Grade II ganglioglioma. Positivity: GFAP, S-100, Leu-7, neurofilament | Total | 6 years. Thrombosis of inferior sagittal sinus. Lumbo-peritoneal valve. Slight delay in school attendance |
| 18 | 15/M | Incidental | – | Right temporal | Grade I ganglioglioma. Positivity: GFAP, NSE | Total | 6 years. Asymptomatic |
| 19 | 2.5/M | Generalized tonic-clonic seizure | 1 month | Left temporal | Grade II ganglioglioma. Positivity: GFAP, NSE, chromogranin, synaptophysin | Subtotal | Relapse after 16 months. Reintervention. 22 months. Demise |
| 20 | 14/M | Simple partial seizure | 4 months | Right precentral | Grade I ganglioglioma. Positivity: GFAP, NSE | Total | 3 years. Isolated crises in relation with intake of toxics (alcohol, drugs). Requires medication |
NSE: neuron-specific enolase; M: male; F: female; GFAP: glial fibrillary acidic protein.
With the exception of one 15-year-old patient whose diagnosis was incidental following a motor vehicle accident, all of the patients debuted with epileptic seizures which were generalized motor in 7 cases, simple partial in 7, and complex partial in 5 subjects, with a time span varying between 1 month and 29 years from the onset of symptoms until the definitive diagnosis. This interval was 1–180 months (mean 25 months) in the paediatric patient population and 1–29 years in adults (mean: 10.3 years).
Neuroradiological diagnosisComputed tomography (CT) was performed on the 20 patients; the tumour was hypodense in 16 cases, hyperdense in 2, and heterogeneous in another one. Patient number 20 presented a study that was deemed normal. In 14 cases, enhancement was between slight and moderate with contrast and in 5 patients, no uptake was observed (Fig. 1).
.")
In T1-weighted sequences, the magnetic resonance imaging (MRI) carried out in all the patients revealed that 12 tumours were presented as hypointense; 6 were hyperintense; 1 was isointense and the remaining tumours were mixed. In T2-weighted sequences, all the gangliogliomas were hyperintense (Fig. 2). In all cases, gadolinium enhancement was between moderate and intense. Twelve tumours were cystic and eight were solid.
 T1-weighted sequence with contrast. (B)–(D) T2-weighted sequences.")
Angiography was only performed in one 2-year old patient (case number 19) with a bulky tumour located on the left temporal area, with a shift in the vascular structures (middle brain) and diffuse tumour staining.
LocationSeventeen tumours (9 right/8 left) were located on the temporal lobe (Figs. 3 and 4) and the other three were located on the occipital lobe (2 cases) with the third in a precentral location.
 and (B) CAT without and with contrast corresponding to a temporal ganglioglioma. (C) and (D) T1-weighted MRI sequences without and with contrast. (E) and (F) T2-weighted MRI sequences (case 17).")
, T2-weighted (B) and FLAIR (C) sequences, T1 without and with contrast (D and E) and echogradient (case 10).")
Electroencephalographic testing (EEG) was carried out in all the patients and different forms of alteration were found in all symptomatic cases; the child whose tumour was an incidental finding presented a normal EEG. In 6 patients, a video-EEG study was conducted and the seizures could be evaluated in 4 cases.
TreatmentAll the patients underwent surgery, using microsurgical techniques. Macroscopically total extirpation was performed in 17 cases and more than 75% of the tumour was removed in the other 3. Patient number 7, in whom an incomplete resection was completed, presented regrowth at 14 months and, following a second intervention, which was also incomplete, radiotherapy was applied.
ComplicationsThere were only two complications worth mentioning. The patient with grade III ganglioglioma presented ventricular dilatation that was resolved by implanting a ventricle-peritoneal valve and one 2-year old child who, 3 months following surgery, developed a tension cyst on the surgical bed and that required implantation of a cysto-peritoneal shunt. Months later, he developed intracranial hypertension due to thrombosis of the inferior sagittal sinus that necessitated implantation of a lumbo-peritoneal shunt after removal of the previous valve.
EvolutionThe mean follow-up time was 8.5 years (range 22 months–14 years). The 5-year disease-free survival rate in the patients with this minimum follow-up was 85% and the global survival rate was 95%. Of the 18 patients who debuted with epilepsy and who were still alive after 2 years, 5 cases (27.7%) did not require anti-seizure medication; this reduction was more significant in the paediatric population (3 cases, 33.3%) than in the adults (2 cases, 18.8%).
Four patients underwent subsequent surgery: in three cases due to regrowth of tumour remains following incomplete resection, while the other case presented relapse 54 months after having performed a resection that was considered both surgically and radiographically complete. In these cases, the tumours were 2 grade II gangliogliomas, one grade III ganglioglioma, and the other one presented an oligodendroglial component. There was only one death (case number 19) due to tumour progression.
HistologyAll the surgical specimens underwent histological study with haematoxylin–eosin and specific immunohistochemical techniques. Eleven cases presented a typical grade I ganglioglioma and in 6 cases, it was a WHO grade II tumour; another case presented a mixed oligodendroglial (grade II) component, and a third tumour was a WHO grade III anaplastic ganglioglioma (Figs. 5 and 6). In the 4 cases who underwent a second surgery, no modification was observed in the type or grade of tumour.
 and neural (arrow) components (HE 100×).")
 Very scant positivity for P53. (B) Positivity for synaptophysin in cells.")
In 4 cases of grade II ganglioglioma, Ki 67 determination was performed and was weakly positive; CD34 determination was not carried out in any of the samples obtained.
DiscussionThe review of the WHO classification encompasses gangliogliomas under the term neural and mixed glioneural tumours, which also includes gangliocytomas, dysplastic gangliocytoma of the cerebellum (Lhermitte–Duclos disease), childhood desmoplastic astrocytoma/gangliocytoma, neuroepithelial disembryoplastic tumour, central neurocytoma, cerebellar liponeurocytoma, and paraganglioma of the filum,3 to which a series of very uncommon tumours have been added, such as glioneural papillary tumour, the rosette-forming glioneural tumour of the fourth ventricle.4
The term ganglioglioma was introduced by Courville in 19305 to describe a mixed tumour with extremely low incidence, in which an astrocytic component and another neural type component were particularly striking. In the general population, they represent between 0.4% and 0.7% of all brain tumours and up to 1% of medullary tumours; however, they account for 5% of all childhood tumours.1
In most of the series, there is a slight preference of males over females2,6 and in our case registry, there is also a 1:1.4 predominance of males over females; however, in the paediatric subseries, all the patients were male, and among the adults there were twice as many women as men. These tumours can present at any age and there are case reports from the age of 2 months and 80 years, although in our experience the mean age is 26.4 years at the time of diagnosis. Incidence appears to be unaffected by geography or race.
In our case series, as in the majority of the series, the most common form of presentation was with long-standing, drug-refractory, epileptic seizures2 and focal neurological deficits are exceptional. In gangliogliomas located in the fossa posterior, as in other tumours located here, the cranial nerves, signs of cerebellar dysfunction, headache, etc. can be found.7 Cases of gangliogliomas associated with other diseases, such as type I and II neurofibromatosis, Down's syndrome, Turcot's syndrome, Peutz–Jeghers’ syndrome, partial agenesia of the corpus callosum, cerebellar polymicrogyria,6,8 have also been reported.
Although Courville's initial description predominantly located GG in the III ventricle and they can, topographically, be located anywhere in the CNS, the vast majority are located in the temporal lobe (17 cases in our series), albeit they are not uncommon in the parietal and frontal lobes.1,9 In our experience, we have not had any gangliogliomas outside the cerebral hemispheres; they have been reported less frequently in the cerebellum,10 brainstem,11 pituitary,12 pineal,13 thalamus,14 and hypothalamus,15 supratentorial and IV ventricles,16 optic and trigeminal nerves,17 suprasellar region,18 ocular globe,19 or the spinal cord.20 The infratentorial location (stem, cerebellum, and cerebellopontine angle) is rare with few cases reported in the literature.21,22 In exceptional cases, they can be bilateral or multicentric.23
Microscopically, they are characterized by the presence of neurons and mature glial cells, generally pilocytic or fibrillary astrocytes and, less often, oligodendrocytes. The neural population is disordered with morphological varieties in terms of size and it is not uncommon to see ganglionary pyramidal cells and/or well differentiated ganglionary cells. Mitoses are uncommon and are seen exclusively in the glial component.24 They may be accompanied by cortical dysplasia with cortical disorganization, microscopic glioneural dysplasias due to alterations of neural migration or present associated with a component of pleomorphic xanthoastrocytoma.25 Exceptionally, malignancy data such as anaplasia, vascular proliferation, and necrosis are observed. Although the WHO grade is only indicated in 9 of our patients (6 were grade II and 3 were grade I), up to 93% of cases in the literature are grade I, 6% are grade II, and a mere 1% are grades III and IV.22
Several different varieties of gangliogliomas have been reported, such as melanotic26 or microcystic,27 as well as gangliogliomas associated with other cerebral neoplasms, such as haemangioma,28 pleomorphic xanthoastrocytoma,29 or with the presence of other neoplastic components, for instance, oligodendroglial components30 as occurred in our patient number 12 or they can present a tanycytic ependymoma component.31
Immunohistochemical techniques are fundamental to identify the two cell components, thus, the glial portion stains positive for protein S-100, glial fibrillary acidic protein, and vimentin, whereas the neural population stains positive for synaptophysin, neuron-specific enolase, chromatogranin A, calbindin, and calcineurin,1,2 up to 80% of all GG stain positive for CD34.2 Some biomarkers such as leptin and its receptor, which are over-expressed in malignant glial tumours, present a low level of expression in others that have a low degree of aggressiveness, such as the ganglioglioma.32 In the last few years, some works have been published that address the molecular genetics of GG with the identification of several mutations that affect chromosomes 9, 10,1,8 and, for the first time, the presence of a mutation on the NBN gene was recently reported33 in a supratentorial GG resected in a 13-year old girl.
In the computed tomography, 40% are hypodense; 30% reveal a hypodense cyst with an isodense mural node; 15% are isodense, and the remaining 15% are hyperdense. Between 20% and 83% present calcifications. In 50% of the cases, they are contrast-enhanced.34
With magnetic resonance imaging, they present as well-delimited masses that in T1 are isodense (40–70%) or hypointense (20–40%), and hyperintense (70–90%) or isointense (20–30%) in T2. They show variable and generally non-uniform enhancement with the administration of gadolinium.24,34 They tend to exert little mass effect, are accompanied by scant oedema, and only rarely are data of old bleeding observed. The presence of vasogenic oedema is characteristic of high grade gangliogliomas and areas of cortical dysplasia can occasionally be seen.
In general, angiographic study is not necessary, given that vascular displacement is the only observation as they have little vascularization.
The radiological differential diagnosis should be made with other intracranial cystic lesions, for instance, dermoid and epidermoid cysts,35 cystic haemangioblastomas,36 and with tumour lesions such as pilocytic astrocytoma or childhood desmoplastic ganglioglioma.24 Cystic, typically bulky, lesions are commonly seen in childhood desmoplastic ganglioglioma that may contain walls in the interior that are usually in contact with the dura mater and that present dural enhancement.24 MRI spectroscopy shows an increase in the peak value for choline and the Choline/N-acetyl aspartate ratio (Cho/NAA), as well as a reduction in the Choline/Creatine (Cho/Cr) and NAA/Cr ratios, in relation to the same area of the contralateral hemisphere when normal or with gliomas.37
Recently, Kikuchi et al.38 studied the apparent diffusion coefficient (ADC) with MRI in a group of 10 consecutive patients with supratentorial ganglioglioma, and compared them with another group of high and low grade astrocytic tumours, observing that the mean coefficient in GG is 1.45±0.20×10−3 mm2/s, significantly higher than that obtained in astrocytomas with different degrees of malignancy. In addition, when comparing the ADC with the cellularity of the gangliogliomas, an inverse correlation is observed between them. In the PET and SPECT studies, low grade gangliogliomas show normal or reduced metabolic activity, whereas hyperactivity is seen in high grade gangliogliomas.39
The first treatment option for these lesions is complete surgical resection whenever possible, in which case no other supplementary treatment would be necessary.40 Low grade gangliogliomas are not very sensitive to radiation so radiotherapy should only be used in cases of relapses that are not suitable for further surgical resection,41,42 where astrocytes are observed to have signs of aggressiveness, or when a malignant degeneration occurs. Nonetheless, since it is a tumoral strain of low malignancy and considering the harmful effects of radiation on the maturity of the infant brain, it is currently difficult to justify. Other negative aspects derived from radiation would be the induction of new brain tumours42 or the possibility of malignant degeneration. However, the literature contains cases that have suffered malignant degeneration without radiotherapy having been administered.2,43
Rades et al.44 recently reviewed the 402 cases of GG published in the literature, whether in isolated cases or in extensive series. They found that 15% (60 cases) were labelled as malignant depending on the presence of a high MIB-1 (>10%) or anaplastic changes in the tumours. In the 342 cases labelled as low grade, only surgical treatment was performed on 268 patients (78.4%) and surgery was complemented with radiotherapy in 74 tumours (14 complete and 60 partial resections); in other words, 21.6% of low grade GG received radiotherapy. They concluded the revision by indicating that complete resection does not require supplementation with radiotherapy and that, in cases of incomplete removal, post-operative radiotherapy ostensibly improves local control of the tumour. This revision by Rades et al.,44 however, included desmoplastic gangliogliomas of both children and adults despite they are being clinically and histopathologically different entities, so the claims made in it must be interpreted with caution.45
There are no conclusive data on the benefits of chemotherapy.46
The prognosis is very favourable when complete resection is performed, with relapse rates of 3% after 2 years. Patients presenting epileptic seizures are free from these in more than 75% of cases, with better control in the rest with specific medication.2,47,48 In the revision by Phi et al.49 analyzing 87 patients with low grade glioma who debuted with epilepsy, post-surgical monitoring of their seizures improved over time, with 79% control being achieved in the first year, growing to 86% in the second and 92% in the fifth year. In infratentorially located tumours, special care must be taken with the possibility of causing neurological deficits and, in the case of any tumour left behind, its behaviour is more benign than that of other tumours in the same location.21 In relapse cases, it is common to see an increase in the degree of malignancy at the expense of the glial component.
The reduction or suppression of anti-epileptic drugs is in general studied little in the literature; in our experience, 5 patients (27.7%) did not require medication 2 years after surgery. In the series of 67 patients with tumour-related epilepsy in the temporal lobe reported by Zaatreh et al.,50 only 17 (28% of the total) out of the 44 patients who remained seizure-free were able to suspend medication entirely. In the paediatric series of Iannelli et al.,51 the percentage of children free of medication was only 16%. Surprisingly, the recent publication by Phi et al.49 shows a percentage of 89%, which the authors attribute to the different statistical methods used, the various monitoring systems used or the different characteristics of the series analyzed as the degree of medication withdrawal is higher in paediatric patients than in adults, although this statement is not supported by other bibliographical references51; another factor to be borne in mind when analyzing this fact is, in our opinion, the heterogeneity of the series published with a variety of ages, locations, tumour types, etc.
The factors considered indicative of good prognosis and therefore with a lower relapse rate are grade I tumours, those located in the temporal lobe, those debuting with long-standing epilepsy and when complete excision is performed and demonstrated by magnetic resonance imaging.2,52–54 The age of the patient at the moment of surgery is of no prognostic value.2
Factors indicating a poor prognosis, apart from higher histological grades, must include the presence of cellular atypia,10,53 location on the midline10 and incomplete resection, although this is not mentioned in the series by Lang et al.55 or Rumana et al.56; it must be remembered that these authors do not mention post-operative MRI studies.
In our series, determination of Ki-67 and p53 was not performed in any case even though these parameters may have an important prognostic value, as shown by Hirose et al.57 when observing significantly higher figures in cases of relapse with respect to no-recurrent cases. Dissemination through cerebrospinal fluid is infrequent. In the series published by Hukin et al.58 analyzing 96 gangliogliomas, they observed 4 cases, of which only two were in the cerebral hemispheres, with the other two in the brainstem and medulla of the spine.
Anaplastic gangliogliomas behave more aggressively at the local level and also cause leptomeningeal seeding, although the existence of this dissemination does not in principle imply, for Hukin et al.,58 a worse prognosis when compared with other tumours of comparable malignancy. In anaplastic or relapsed gangliogliomas, evidence of “anti-apoptotic survivin” protein in more than 5% of glial cells, shown through immunohistochemical techniques, is considered to be a poor prognosis factor.59
As our study is limited by the number of cases, we can conclude that it is not possible to conduct a rigorous statistical study.
Conflicts of interestThe authors have no conflict of interests to declare.
Please cite this article as: Gelabert-González M, et al. Gangliogliomas intracraneales. Revisión de una serie de 20 pacientes. Neurología. 2011;26:405–15.
