



La neurofibromatosis tipo 1 (NF1) es un desorden progresivo multisistémico de herencia autosómica dominante que presenta numerosas manifestaciones neurológicas.
MétodosRevisión de historias clínicas de pacientes afectos de NF1 controlados en una Unidad de Neuropediatría de mayo de 1990 a 31 de diciembre de 2018 y sus manifestaciones neurológicas asociadas.
ResultadosSe revisaron 128 pacientes afectos de NF1. Edad media al diagnóstico de NF1, 4,43 años±3,38 SDS (rango 6 meses-14,5 años) con discreto predominio femenino (53,1%). Se asocia macrocefalia (PC>2SDS) en el 37,5% de los casos. TDAH en el 28,9% de los casos (37), subtipo combinado 20, inatento 15 casos y predominantemente hiperactivo 2 casos. Otras manifestaciones incluyen; cefalea (18,7%), déficit cognitivo (7,8%), afectación motora (6,2%) y epilepsia (4,68%). Se realizó RM cerebral a 85 pacientes, mostrando 60 (70,5%) hiperseñales en T2 en ganglios basales y/o cerebelo, junto con otras alteraciones como Chiari I (4 casos) y quistes aracnoideos (3 casos). Se identificaron gliomas de nervio óptico en 22 casos (25,8%). Otros hallazgos diagnosticados por RM incluyen neurofibromas plexiformes (9,3%) y otros gliomas localizados en sistema nervioso central (3,1%).
ConclusionesLas manifestaciones neurológicas encontradas concuerdan con lo recogido en la literatura. El seguimiento de estos pacientes se pierde en la edad adulta, siendo necesario establecer adecuadas estrategias de transferencia y posterior seguimiento de pacientes a los servicios de adultos.
Neurofibromatosis type 1 (NF1) is a progressive multisystem disorder following an autosomal dominant inheritance pattern that presents with multiple neurological manifestations.
MethodsWe reviewed medical histories of patients with NF1 followed up at our hospital's paediatric neurology department from May 1990 to 31 December 2018. We collected data on neurological symptoms.
ResultsA total of 128 patients with NF1 were identified. Mean age (SD) at NF1 diagnosis was 4.43 (3.38) years (range, 0.5-14.5 years). There was a slight female predominance (53.1%). Macrocephaly (head circumference over 2 SDs above average for age) was present in 37.5% of cases. Attention-deficit/hyperactivity disorder was recorded in 28.9% of patients (37): combined type in 20 patients, predominantly inattentive in 15, and predominantly impulsive/hyperactive in 2. Other manifestations included headache (18.6%), cognitive impairment (7.8%), motor deficit (6.2%), and epilepsy (4.68%). Brain MRI was performed in 85 patients, revealing T2-weighted hyperintensities in the basal ganglia and/or cerebellum in 60 patients (70.5%), Chiari malformation type 1 in 4 cases, and arachnoid cysts in 3. Optic nerve gliomas were identified by MRI in 22 patients (25.8%). Other MRI findings included plexiform neurofibromas (9.3%) and central nervous system gliomas (3.1%).
ConclusionsThe neurological manifestations identified in our sample are consistent with those reported in the literature. Effective transfer strategies from paediatric neurology departments and subsequent clinical follow-up by adult neurology departments are needed to prevent loss to follow-up in adulthood.
La neurofibromatosis tipo 1 (NF1) es un desorden progresivo multisistémico de herencia autosómica dominante, con una elevada tasa (50%) de mutaciones espontáneas. Se estima una prevalencia de 1/2500-3000 individuos1.
Es causada por mutaciones en el gen NF1 localizado en el cromosoma 17q11.2. El producto del gen NF1, llamado neurofibromina, se expresa de forma ubicua, por lo que las complicaciones pueden afectar a prácticamente cualquier parte del organismo en cualquier momento de la vida, especialmente aparición y crecimiento de tumores nerviosos junto con otras anomalías como alteraciones cutáneas y óseas. En el pasado el diagnóstico se establecía en base a criterios clínicos. Actualmente se identifican mutaciones en más del 95% de los casos. Existe una gran variabilidad clínica y escasa correlación genotipo-fenotipo1.
Las complicaciones neurológicas constituyen uno de los hallazgos más frecuentes2. Los problemas de aprendizaje y del lenguaje junto con trastornos de inatención, hiperactividad y/o impulsividad se dan en el 30-60% de los casos3. Otras complicaciones neurológicas incluyen epilepsia, generalmente secundaria a lesión estructural, cefalea de carácter migrañoso, gliomas cerebrales, malformaciones intracraneales, aneurismas y síndrome de Moya-Moya entre otros2.
La resonancia magnética (RM) es la técnica de elección para la identificación de alteraciones como gliomas de nervio óptico, tumores cerebrales, y malformaciones4. Un elevado porcentaje de los pacientes presentan aumentos de señal en secuencias ponderadas en T2, a nivel de tractos ópticos, cerebelo, troncoencéfalo, tálamo, globo pálido y cápsula interna, hallazgos conocidos como «imágenes brillantes inespecíficas» (IBI o su pseudónimo en inglés UBO), de significado clínico incierto y que tienden a desaparecer con la edad4.
Se revisan los pacientes afectos de NF1 controlados desde mayo de 1990 en la sección de Neuropediatría y sus manifestaciones neurológicas asociadas.
MétodosEn la sección de Neuropediatría de nuestro centro se trabaja con protocolos clínicos, hojas de información y una base de datos con los niños valorados desde mayo de 19905. Desde agosto de 2012 se entrega la hoja informativa sobre «Neurofibromatosis 1». En ella se explica en qué consiste esta entidad así como las pruebas diagnósticas y controles periódicos que deben seguir estos pacientes, de acuerdo con nuestro protocolo establecido siguiendo las guías de práctica clínica. Entre el seguimiento a realizar se encuentra valoración clínica anual o semestral en menores de 2 años, examen oftalmológico anual hasta los 8 años y bienal hasta los 18 años, examen dermatológico anual, exploración física exhaustiva en busca de alteraciones óseas, control de tensión arterial anual especialmente en adolescentes y vigilancia de problemas de aprendizaje entre otros6,7. (Anexo 1).
Se realiza un estudio observacional, retrospectivo y descriptivo mediante revisión de las historias clínicas de los pacientes afectos de NF1 que figuran en la base de datos de la Unidad de Neuropediatría desde mayo de 1990 hasta el 31 de diciembre de 2018, y sus manifestaciones neurológicas identificadas.
Este trabajo fue aprobado por el Comité Ético de Investigación Clínica de Aragón con el número de expediente PI16/092.
ResultadosEn la base de datos de Neuropediatría desde mayo de 1990 al 31 de diciembre de 2018 constaban 136 pacientes afectos de NF1, de un total de 21789 pacientes incluidos. Se revisaron las historias clínicas de 128 pacientes afectos de NF1, ya que en 8 casos, no se pudieron conseguir las historias clínicas por no estar presentes en los archivos clínicos del hospital dado el largo tiempo de ausencia de seguimiento. De ellos 97 pacientes presentaban diagnóstico genético de NF1 y 31 casos diagnóstico por criterios clínicos, por ser casos antiguos en los que no se realizó genética o las técnicas utilizadas no identificaban todas las mutaciones.
La edad media de los pacientes al diagnóstico de NF1 fue de 4,43 años±3,38 SDS (rango 6 meses-14,5 años), existiendo discreto predominio femenino (53,1%): 60 niños y 68 niñas. El tiempo medio de seguimiento fue de 6,92 años±4,90 SDS (rango 1 mes-20,9 años). La edad media de la última visita es de 10,99 años±5,35 SDS (rango 1 año-36,5 años; uno de los pacientes, no controlado desde los 20 años, acudió a los 36 años porque su hijo de 5 meses presentaba manchas café con leche)8.
Actualmente siguen en control 50 pacientes, 34 mujeres (68%) y 16 hombres (32%) con una edad media de 9,27años±4,16 SDS, y con un rango de edad entre año y medio y 17 años y medio.
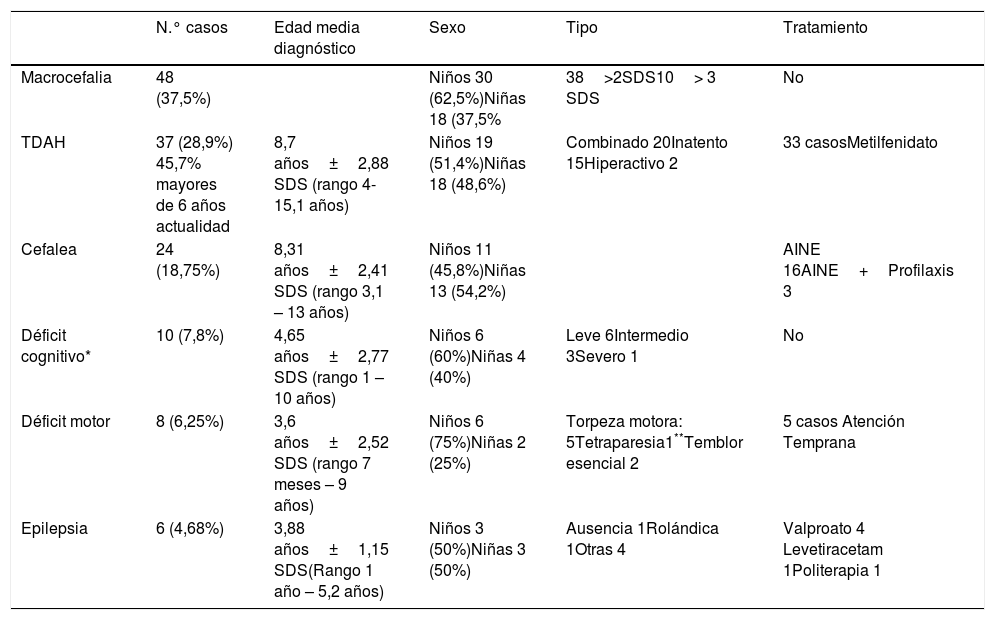
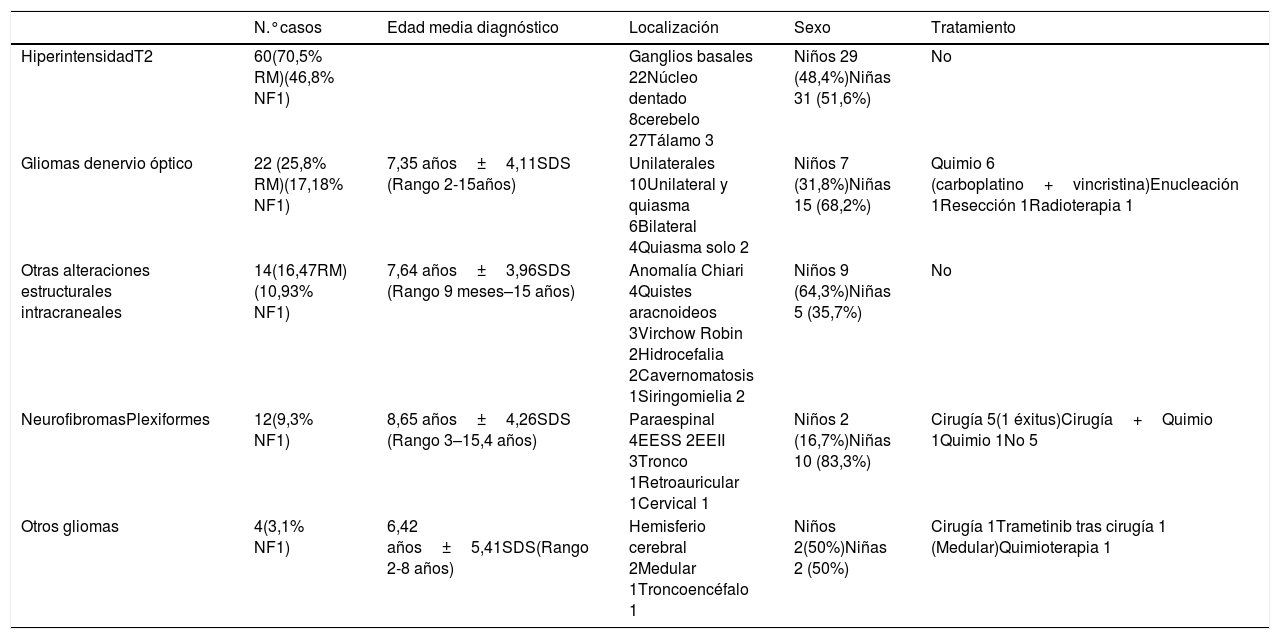
Las manifestaciones neurológicas, los hallazgos de RM en los casos realizados y los tratamientos empleados se han recogido en las tablas 1 y 2.
Manifestaciones neurológicas en pacientes con NF1
| N.° casos | Edad media diagnóstico | Sexo | Tipo | Tratamiento | |
|---|---|---|---|---|---|
| Macrocefalia | 48 (37,5%) | Niños 30 (62,5%)Niñas 18 (37,5% | 38>2SDS10> 3 SDS | No | |
| TDAH | 37 (28,9%) 45,7% mayores de 6 años actualidad | 8,7 años±2,88 SDS (rango 4-15,1 años) | Niños 19 (51,4%)Niñas 18 (48,6%) | Combinado 20Inatento 15Hiperactivo 2 | 33 casosMetilfenidato |
| Cefalea | 24 (18,75%) | 8,31 años±2,41 SDS (rango 3,1 – 13 años) | Niños 11 (45,8%)Niñas 13 (54,2%) | AINE 16AINE+Profilaxis 3 | |
| Déficit cognitivo* | 10 (7,8%) | 4,65 años±2,77 SDS (rango 1 – 10 años) | Niños 6 (60%)Niñas 4 (40%) | Leve 6Intermedio 3Severo 1 | No |
| Déficit motor | 8 (6,25%) | 3,6 años±2,52 SDS (rango 7 meses – 9 años) | Niños 6 (75%)Niñas 2 (25%) | Torpeza motora: 5Tetraparesia1**Temblor esencial 2 | 5 casos Atención Temprana |
| Epilepsia | 6 (4,68%) | 3,88 años±1,15 SDS(Rango 1 año – 5,2 años) | Niños 3 (50%)Niñas 3 (50%) | Ausencia 1Rolándica 1Otras 4 | Valproato 4 Levetiracetam 1Politerapia 1 |
Alteraciones estructurales en RM
| N.°casos | Edad media diagnóstico | Localización | Sexo | Tratamiento | |
|---|---|---|---|---|---|
| HiperintensidadT2 | 60(70,5% RM)(46,8% NF1) | Ganglios basales 22Núcleo dentado 8cerebelo 27Tálamo 3 | Niños 29 (48,4%)Niñas 31 (51,6%) | No | |
| Gliomas denervio óptico | 22 (25,8% RM)(17,18% NF1) | 7,35 años±4,11SDS (Rango 2-15años) | Unilaterales 10Unilateral y quiasma 6Bilateral 4Quiasma solo 2 | Niños 7 (31,8%)Niñas 15 (68,2%) | Quimio 6 (carboplatino+vincristina)Enucleación 1Resección 1Radioterapia 1 |
| Otras alteraciones estructurales intracraneales | 14(16,47RM)(10,93% NF1) | 7,64 años±3,96SDS (Rango 9 meses–15 años) | Anomalía Chiari 4Quistes aracnoideos 3Virchow Robin 2Hidrocefalia 2Cavernomatosis 1Siringomielia 2 | Niños 9 (64,3%)Niñas 5 (35,7%) | No |
| NeurofibromasPlexiformes | 12(9,3% NF1) | 8,65 años±4,26SDS (Rango 3–15,4 años) | Paraespinal 4EESS 2EEII 3Tronco 1Retroauricular 1Cervical 1 | Niños 2 (16,7%)Niñas 10 (83,3%) | Cirugía 5(1 éxitus)Cirugía+Quimio 1Quimio 1No 5 |
| Otros gliomas | 4(3,1% NF1) | 6,42 años±5,41SDS(Rango 2-8 años) | Hemisferio cerebral 2Medular 1Troncoencéfalo 1 | Niños 2(50%)Niñas 2 (50%) | Cirugía 1Trametinib tras cirugía 1 (Medular)Quimioterapia 1 |
La macrocefalia constituyó el hallazgo más frecuente, y de predominio en varones.
La cefalea se ha asociado con alteraciones estructurales en 11 casos: dos anomalías de Chiari, dos afectos por siringomielia, 1 anomalía de Virchow Robin, cuatro gliomas ópticos y dos gliomas cerebrales.
La discapacidad intelectual se dio en 10 pacientes, generalmente de carácter leve (valorada por la escala Wechsler: WPPS-III Preescolar y primaria y WISC-IV para niños considerando retraso mental leve entre 70-55, moderado 55-40, y severo 40-25). Cinco de estos pacientes presentaban de forma asociada alguna forma de epilepsia.
Se realizó RM cerebral a 85 pacientes (66,4%). Entre los hallazgos encontrados predominaron las hiperintensidades en secuencia T2, gliomas de nervio óptico y alteraciones estructurales intracraneales. Otros hallazgos identificados por resonancia magnética son los neurofibromas plexiformes y la presencia de gliomas en otras localizaciones (tabla 2).
Los gliomas ópticos predominaron en el sexo femenino (68,2%), identificados por RM como hallazgo casual en 13 casos y tras indicación por oftalmología por hallazgos en la exploración como edema de papila, 4 casos y atrofia papilar, 5 casos. Cuatro casos presentaron síntomas previos a la exploración oftalmológica, tres de ellos asociando proptosis ocular junto con pérdida de agudeza visual y una paciente déficit visual aislado. Se trataron 9 casos, quimioterapia en 6 casos, con combinación de carboplatino y vincristina. En un paciente se realizó enucleación del ojo izquierdo en 2015 debido a importante extensión tumoral con afectación papilar a pesar de tratamiento con quimioterapia. Una paciente fue sometida a exéresis tumoral retrobulbar con secuela de amaurosis de ojo derecho a los 4 años de edad (1994) y otra paciente fue sometida a corticoterapia inicial seguida de radioterapia en el año 2009 con regresión parcial de tumoración. El resto de los pacientes no han precisado tratamiento, sin presentar cambios en la evolución del glioma óptico.
Un paciente no controlado desde la edad pediátrica con glioma óptico derecho y glioma de cápsula interna izquierda, acudió a la edad de 36,5 años a la consulta tras derivación de su hijo de 5 meses con manchas café con leche. Se objetivó en una RM solicitada por su médico de Atención Primaria en 2012 (tras 13 años sin control), una regresión total de glioma óptico derecho y una disminución de tamaño de glioma de cápsula interna izquierda8.
Otras alteraciones estructurales supusieron en la mayor parte de las ocasiones hallazgos incidentales, en su mayoría asintomáticos. Se objetivó dilatación ventricular en 2 pacientes varones, ambos asintomáticos. En el primer caso la hidrocefalia se cree motivada por la presencia de una lesión hiperintensa en T2 a nivel parasagital izquierdo, que no llegaba sin embargo, a producir obstrucción ni desplazamiento de línea media, y en el segundo caso a la presencia de múltiples hiperintensidades que provocaban discreta estenosis de acueducto de Silvio. Ninguno de los dos precisó tratamiento de hidrocefalia tras dos años y medio y trece años de seguimiento respectivamente.
El caso de cavernomatosis asociada, se debe a la mutación responsable de cavernomatosis múltiple familiar p.E166X (c.496G>T) en el exón 8 del gen PDCD10 (CCM3), heredada del padre. La mutación responsable de NF1 había sido heredada de la madre.
Los neurofibromas plexiformes fueron más frecuentes en mujeres (83,3%), siendo la aparición de un bultoma en el área afecta la forma más frecuente de manifestación. Tres pacientes, presentaron clínica de dolor acompañada de otras manifestaciones como cifosis dorsal en un caso, por presencia de neurofibroma plexiforme dorsal y 2 casos claudicación y limitación de la marcha por presencia de neurofibromas plexiformes paraespinales lumbares. Un caso fue éxitus a la edad de 12 años debido al crecimiento exponencial de un neurofibroma plexiforme localizado en mediastino, recidivante a pesar de varias resecciones quirúrgicas.
La presencia de otros gliomas a nivel cerebral o de otras localizaciones fue un hallazgo incidental al realizar la RM. Dos casos por su localización y tamaño, aunque asintomáticos, fueron tratados con resección quirúrgica de glioma troncoencefálico y tratamiento de lesión medular con Trametinib (inhibidor de la proteína MEK) como uso compasivo, debido a la imposibilidad de realizar una resección quirúrgica completa tras resección parcial del mismo. En el caso de los gliomas cerebrales localizados en hemisferios, un caso de localización hipotálamica se trató con quimioterapia por clínica de síndrome diencefálico, mientras que en el otro caso se mantuvo una actitud expectante al no objetivarse crecimiento tras 9 años de seguimiento.
DiscusiónLa macrocefalia, considerando un perímetro craneal superior a 2 desviaciones estándar, constituye el hallazgo más frecuente en nuestra muestra: 37,5%, aunque queda algo lejos del 50-75% de casos que describen algunos autores9–11. Algunos la consideran debida a la expansión de algunas estructuras cerebrales, principalmente hemisferios, que daría lugar a megaencefalia12. Este crecimiento parecería influir y estar relacionado con los trastornos de aprendizaje, déficit de atención13 y crecimiento de tumores de vías ópticas14.
El diagnóstico de TDAH se ha establecido en aproximadamente el 30% de nuestros casos, y afecta al 45,7% de los niños en edad escolar controlados en la actualidad; cifras que han ido en aumento en los últimos años. En la literatura se dan cifras de entre 38 y 58,33% de los pacientes NF13,15. El diagnóstico de TDAH en nuestros casos se estableció a una edad media de 8,7 años±2,88 SDS, con un rango de edades de 4 a 15 años similar a los referido en la literatura: media 10,7±2,2 SDS años y rango de 4 a 15 años16. El subtipo clínico combinado (déficit de atención e hiperactividad) es el más frecuente (alrededor del 50%)17, tal y como se da en nuestra muestra, aunque algunos autores discrepan, considerando una mayor prevalencia del subtipo inatento18. El tratamiento con metilfenidato se ha postulado como eficaz en pacientes NF1; en nuestra muestra 33 (89,1%) pacientes fueron tratados18,19.
La cefalea es relativamente frecuente en estos pacientes, aunque nuestros resultados (18,75%), difieren con lo aportado por algunas series que refieren hasta el 50-60% de los pacientes respecto a grupo control20. Es importante en estos pacientes descartar le presencia de alguna complicación como causa de la cefalea, en nuestra muestra presentes en 11 casos20.
La discapacidad intelectual se da con mayor prevalencia en los niños NF1 comparados con la población general; aproximadamente un 6-7% presentan un coeficiente intelectual inferior a 7021. Estos valores son similares a lo obtenido en nuestro estudio (7,8%) donde predomina la discapacidad intelectual leve. Algunos autores han relacionado la discapacidad intelectual con la presencia de epilepsia en pacientes NF122; la mitad de los pacientes con discapacidad intelectual (5 casos) de la muestra presentaban asociada alguna forma de epilepsia.
Las crisis convulsivas afectan alrededor del 6,5-9,5%21 de pacientes NF1, algo superior a lo obtenido en nuestros datos (epilepsia 4,68%). De inicio en la infancia como en nuestro caso (edad media diagnóstico 3,88 años±1,15 SDS) y adolescencia aunque pueden aparecer a cualquier edad23. Predominan las crisis focales, aunque pueden aparecer cuadros bien definidos como epilepsia-ausencias, síndrome de West entre otros24. Por lo general la respuesta al tratamiento antiepiléptico es adecuada, aunque existen casos refractarios y que pueden precisar cirugía22,25; en nuestro caso solo un paciente precisó la combinación de varios fármacos.
La RM es la técnica de elección para el diagnóstico de las alteraciones estructurales intracraneales. Existe controversia acerca de su indicación, algunos autores, consideran que debe realizarse al momento del diagnóstico ya que puede aportar datos al mismo, mientras que otros, consideran que debe realizarse cuando existan signos sugestivos de anomalías intracraneales26. Actualmente las diferentes guías de práctica clínica establecen que la RM no se recomienda de forma rutinaria y debe realizarse cuando exista sintomatología sugestiva de complicaciones o por la identificación de una deleción completa del gen NF1 que se asocia con frecuencia a anomalías cerebrales estructurales4. En nuestro caso se realiza cuando existen manifestaciones clínicas sugestivas de alguna lesión intracraneal y/o medular.
Uno de los hallazgos principales en los niños NF1 son las hiperintensidades en secuencias T2 en la RM cerebral, principalmente a nivel de los ganglios basales y cerebelo, que se encuentran en el 70,5% de las RM realizadas en nuestra muestra, lo que concuerda con las series con valores que oscilan 50% y el 90%4. Responden a áreas de vacuolización intramielínica27, más frecuentes en edades precoces y con un significado clínico incierto4. Algunos estudios, los relacionan con la aparición de trastornos de aprendizaje y deterioro cognitivo28 sin existir datos concluyentes29.
Los gliomas de nervio óptico son los tumores más frecuentes del sistema nervioso central en pacientes NF1, con una prevalencia del 15-25%30comparable a los datos recogidos (17,18%). Generalmente son asintomáticos clínicamente, más frecuentes en menores de 6 años (7,35±4,11 SDS de edad media al diagnóstico en nuestro caso), pudiendo tener a partir de los 6-7 años de edad un lento crecimiento e incluso presentar regresión espontánea8,31. La actitud es expectante con controles periódicos en casos asintomáticos. En casos sintomáticos puede ser precisa la quimioterapia, generalmente tratamiento de primera elección, con la combinación de fármacos como carboplatino/cisplatino, vincristina o un inhibidor de la proteína MEK. La radioterapia debe evitarse, quedando limitada su utilización tras agotar segunda y tercera línea de tratamiento con quimioterapia o inhibidores de MEK.
La cirugía se reserva para aquellos casos con gran sintomatología como proptosis, afectación estética importante y pérdida visual30,32.
Aquellos pacientes con glioma de vías ópticas presentan mayor probabilidad (20%) de presentar gliomas de bajo grado, generalmente astrocitomas pilocíticos o pilomixoides en otras localizaciones del sistema nervioso central30. Representan un 2-3% de los pacientes NF1, cifras semejantes a las objetivadas en nuestro trabajo (3,1%).
Otras alteraciones estructurales intracraneales están presentes en aproximadamente un 5% de los pacientes NF133,34, aunque en nuestra muestra este porcentaje es ligeramente superior (10,9%).
La anomalía de Chiari tipo I, ha sido encontrada en el 5% de la población NF134, generalmente de carácter asintomático, aunque en algunas ocasiones puede ir asociada a otras manifestaciones como cefalea y dolor cervical35.
Los quistes aracnoideos pueden aparecer en pacientes NF1 aunque su prevalencia es difícil de determinar; generalmente asintomáticos, pueden producir síntomas de hipertensión endocraneal, crisis convulsivas y alteraciones visuales por compresión en caso de rotura o crecimiento36,37.
Si bien la presencia de cavernomas no se ha relacionado con la NF1 algunos autores han intentado establecer una relación entre estas entidades en los últimos años34,38. En nuestro caso la asociación claramente es casual, por una mutación heredada del padre (cavernomatosis) y la otra de la madre (NF1).
La hidrocefalia es poco frecuente (1,5%) pero similar a los datos obtenidos por otros autores que estiman una incidencia entre el 1 y 5%39. Generalmente se debe a estenosis del acueducto de Silvio por tumores como es uno de nuestros casos, o patología de base de cráneo40.
Los neurofibromas plexiformes, aparecen con frecuencia mayor (20%) de la encontrada en nuestro estudio (9,3%). Pueden pasar desapercibidos y ser asintomáticos, fundamentalmente en la edad pediátrica, ya que su crecimiento es mayor a partir de la adolescencia, lo que puede retrasar el diagnóstico2; en nuestro caso la edad media aproximada es de 8 años y medio al diagnóstico. En el caso de ser sintomáticos, pueden ser incapacitantes y requerir tratamiento con cirugía41, como es el caso de 6 de nuestros pacientes y/o nuevas terapias como es el caso de Selumetinib, un inhibidor selectivo de la quinasa MEK 1 y 2 que parece inducir la regresión tumoral y que ha evidenciado en ensayo clínico la disminución de tamaño de los neurofibromas plexiformes en pacientes NF142.
El seguimiento de nuestros pacientes se pierde por no haber una consulta específica de adultos con NF1. Debería asegurarse el control de por vida de la patología crónica, especialmente en enfermedades hereditarias y raras. Es necesario establecer adecuadas estrategias de transferencia y posterior seguimiento de pacientes a los servicios de adultos. Es de una enorme responsabilidad la adecuada información y vigilancia de los riesgos asociados, así como del asesoramiento genético y opciones de diagnóstico prenatal. El seguimiento a largo plazo y la adecuada comunicación entre especialistas de niños y de adultos son herramientas de primer orden para el mejor manejo de nuestros pacientes que así se pueden beneficiar de los continuos avances técnicos, científicos y sociales, y para el conocimiento de la evolución natural de las enfermedades.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

