



Meningeal siderosis is a rare disease caused by haemosiderin deposition secondary to recurrent bleeding in the subpial space. This deposition causes progressive onset of glial proliferation, fibrosis, and finally neuronal damage on the surface of the brain. Tumour necrosis factor receptor–associated periodic syndrome (TRAPS) is an autosomal dominant autoinflammatory disease characterised by prolonged episodes of fever, migratory myalgia and exanthema, aseptic serositis, and increased levels of acute-phase reactants.
We present the case of a 46-year-old man with moderate intellectual disability (65%) of unknown origin. His father had presented secondary amyloidosis, and was a heterozygous carrier of the TNFRSF1A variant p.Arg92Gln. The patient had personal history of seizures since the age of 15 years, currently requiring no treatment, and bilateral presbycusis (64% in the right ear and 37% in the left). He consulted due to progressive instability of 2 years’ progression, premature ageing, severe abasia without slowness or difficulties turning, disorientation in time and space, and frequent mood swings. He presented positive Romberg sign, with no dysarthria, swallowing alterations, diplopia, nystagmus, signs of long-tract involvement, or limb dysmetria. All blood and biochemistry analyses, including a total protein test and plasma acute-phase reactant levels, yielded normal or negative results. Analysis of the TTR gene detected no pathogenic variants.
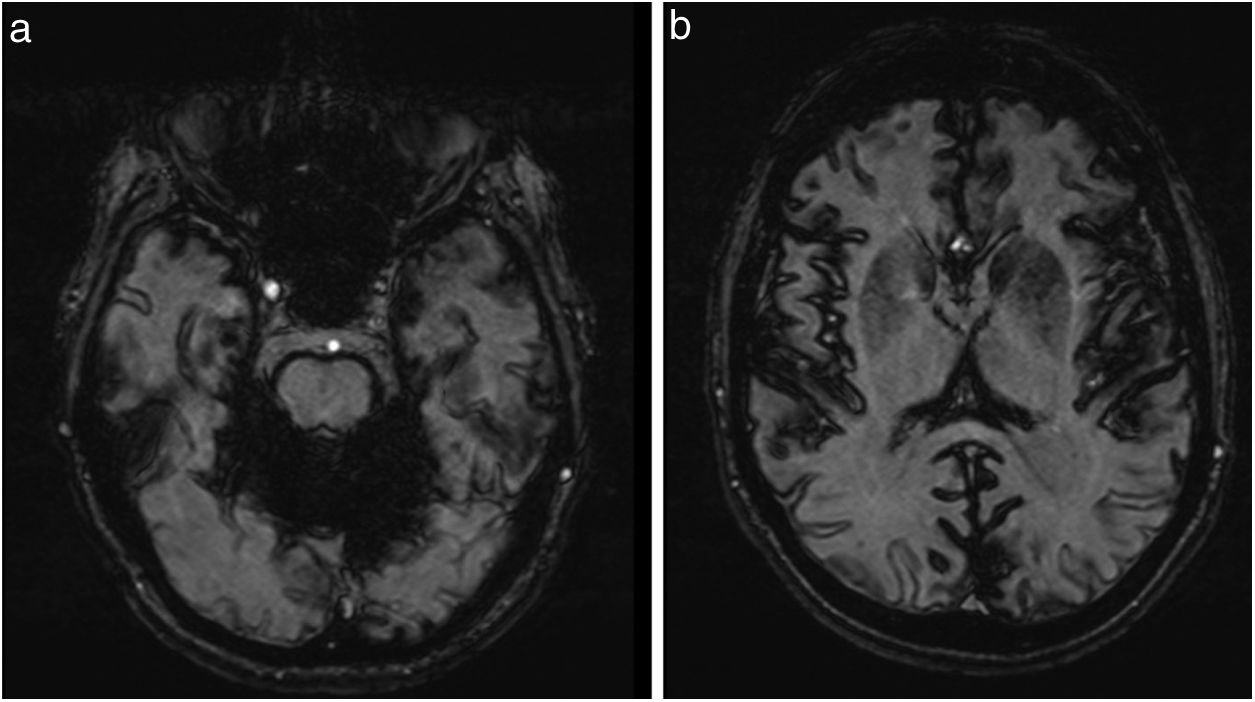
Abdominal ultrasound and colonoscopy findings were normal, and gastroscopy identified hiatus hernia. Duodenal and rectal biopsies did not detect amyloid deposits. Brain MRI revealed cerebellar atrophy, particularly affecting the vermis, and diffuse deposition of haemosiderin in the posterior fossa (Fig. 1A) and, to a lesser extent, in the cerebral sulci (Fig. 1B), with marked magnetic susceptibility in gradient-echo sequences. Electromyography findings were normal.
 Gradient-echo sequences revealed pronounced magnetic susceptibility in the posterior fossa due to diffuse haemosiderin deposition. B) T2-weighted sequences revealed linear hypointensities along the surfaces of the cerebral leptomeninges.")
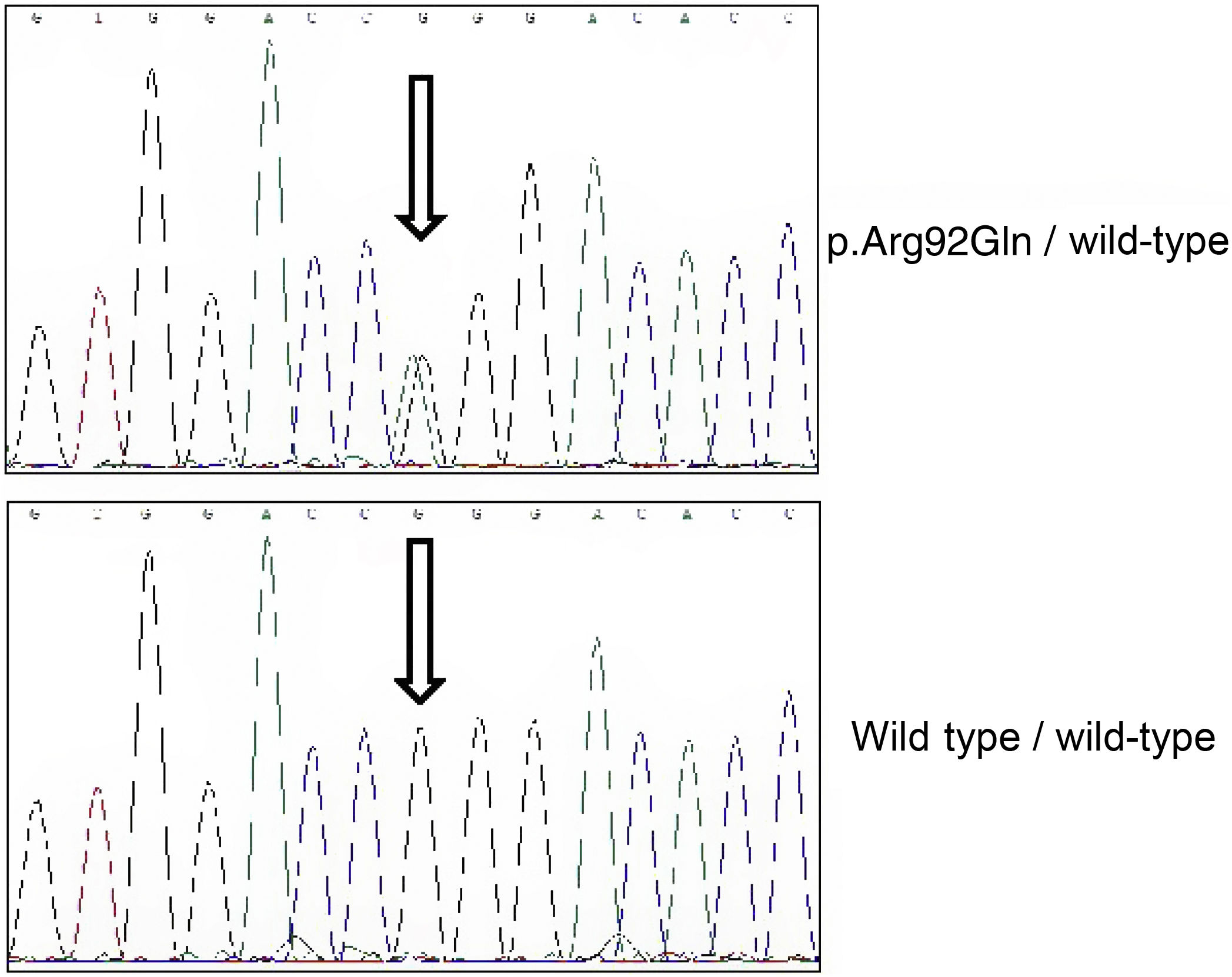
Due to the patient’s family history, we conducted a genetic analysis with a panel of genes involved in autoinflammatory diseases, including the MEFV, TNFRSF1A, MVK, NLRP3, NOD2, PSTPIP1, and CECR1 genes. The library was generated by amplification of all exons and adjacent intronic regions. Sequencing was performed with the NextSeq platform (Illumina), and analysis parameters included minimum coverage of 50×. Sanger sequencing was used to confirm all variants classified as pathogenic, probably pathogenic, or of uncertain significance. The study revealed simple heterozygosis for the p.Arg92Gln variant (Fig. 2).

Meningeal siderosis is a rare disease. Before the development of MRI, its diagnosis was incidental, detected either in surgical procedures or in post mortem examinations. Today, MRI findings are pathognomonic, enabling diagnosis of the condition even at early stages. The most characteristic findings are linear hypointensities along the surfaces of the vermian and cerebellar leptomeninges on T2-weighted sequences, with less extensive involvement of the cerebral leptomeninges.
While the cause is not identified in 50% of patients,1 in other cases the disease is attributed to brain haemorrhage associated with tumours, amyloid angiopathy,2,3 and vascular malformations.4,5 Our patient did not present any of these aetiologies, including leptomeningeal amyloidosis, a rare form of presentation of familial TTR amyloidosis.6 We detected a variant of the TNFRSF1A gene. Mutations in this gene cause TRAPS, an autosomal dominant autoinflammatory disease characterised by prolonged episodes of fever, migratory myalgia and exanthema, aseptic serositis, and increased levels of acute-phase reactants.7 Occasionally, the disease is complicated by AA amyloidosis in adult patients.8
The interest of this case lies in the copresence of meningeal siderosis and TNFRSF1A variant p.Arg92Gln. Firstly, the patient did not present recurrent febrile episodes, nor was there evidence of AA amyloid deposition. TRAPS usually presents in childhood, although it may appear in adulthood in some carriers of the p.Arg92Gln variant.9 Finally, patients with this variant constitute a clinically heterogeneous group, ranging from asymptomatic/presymptomatic individuals to patients who require biological anti-inflammatory treatments.
A previous study demonstrated the presence of microangiopathic changes in the brain of a patient with TRAPS.10 Despite this, there is no clear evidence on the causes of the relationship between meningeal siderosis and TRAPS. One potential explanation may be disruption of blood-brain barrier (BBB) permeability. TRAPS is characterised by a marked increase in levels of multiple circulating inflammatory cytokines. Interleukin-1β plays such an important role that treatments to block this cytokine currently constitute the most effective approach to treating the syndrome.11 In the BBB, increased interleukin-1β levels cause an increase in monocyte chemoattractant protein-1, which may alter the tight junctions of the BBB, increasing its permeability12 and altering the transport of transferrin across the barrier; this would result in ferritin deposition in the brain tissue.
In conclusion, we present the first reported case of a patient with meningeal siderosis and the TNFRSF1A variant p.Arg92Gln. Due to the possible association between the 2 entities, we believe it would be beneficial to include this monogenic autoinflammatory disease in the differential diagnosis of meningeal siderosis.
FundingThis study received no funding of any kind.
Please cite this article as: Nicolás-Sánchez FJ, Aróstegui-Gorospe JI, Piñol Ripoll G, Ribes Amorós I, Nicolás-Sarrat FJ, Sarrat-Nuevo RM, et al. Siderosis meníngea en un paciente portador de la variante p.Arg92Gln del gen TNFRSF1A. Neurología. 2022;37:237–239.
