





Duchenne muscular dystrophy (DMD) is a severe X-linked recessive neuromuscular disease that affects one in 3500 live-born males. The total absence of dystrophin observed in DMD patients is generally caused by mutations that disrupt the reading frame of the DMD gene, and about 80% of cases harbour deletions or duplications of one or more exons.
MethodsWe reviewed 284 cases of males with a genetic diagnosis of DMD between 2007 and 2014. These patients were selected from 8 Spanish reference hospitals representing most areas of Spain. Multiplex PCR, MLPA, and sequencing were performed to identify mutations.
ResultsMost of these DMD patients present large deletions (46.1%) or large duplications (19.7%) in the dystrophin gene. The remaining 34.2% correspond to point mutations, and half of these correspond to nonsense mutations. In this study we identified 23 new mutations in DMD: 7 large deletions and 16 point mutations.
ConclusionsThe algorithm for genetic diagnosis applied by the participating centres is the most appropriate for genotyping patients with DMD. The genetic specificity of different therapies currently being developed emphasises the importance of identifying the mutation appearing in each patient; 38.7% of the cases in this series are eligible to participate in current clinical trials.
La distrofia muscular de Duchenne (DMD) es una enfermedad neuromuscular grave que afecta a uno de cada 3.500 varones nacidos y sigue un patrón de herencia ligada al cromosoma X. En esta enfermedad se observa una ausencia total de la distrofina, generalmente debida a mutaciones en el gen DMD, que altera la pauta de lectura y en torno al 80% de los casos son debidos a deleciones y duplicaciones de uno o más exones.
MétodosSe han revisado 284 casos de varones diagnosticados genéticamente de DMD entre los años 2007 y 2014. Estos pacientes provienen de 8 hospitales españoles de referencia que cubren la mayor parte del territorio español. Para la identificación de las mutaciones se realizaron las técnicas de reacción en cadena de la polimerasa multiplex, MLPA y secuenciación.
ResultadosLos pacientes con DMD presentan en su mayoría grandes deleciones (46,1%) o grandes duplicaciones (19,7%) en el gen de la distrofina. El restante 34,2% corresponde al conjunto de mutaciones puntuales, destacando las sustituciones nucleotídicas tipo nonsense que aparecen en la mitad de los casos. Este estudio permitió identificar 23 nuevas mutaciones en DMD: 7 grandes deleciones y 16 mutaciones puntuales.
ConclusionesEl algoritmo de diagnóstico genético aplicado por los centros participantes es el más adecuado para genotipificar a los pacientes con DMD. La especificidad genética de las distintas terapias en desarrollo pone de manifiesto la importancia de conocer la mutación de cada paciente, siendo un 38,7% de ellos susceptibles de participar en los ensayos clínicos actuales.
Dystrophinopathies constitute a group of neuromuscular diseases caused by alterations of the gene responsible for dystrophin protein (DMD). This gene, which is one of the largest known in humans (79 exons and approximately 2.2 Mb of genomic DNA1), is located in region Xp21; these diseases therefore display an X-linked inheritance pattern. Dystrophin is a 427kDa muscular protein that plays a fundamental role in stabilising the sarcolemma. It does so by using a complex of glycoproteins associated with dystrophin to anchor actin filaments within the cytoskeleton and the extracellular matrix. Lack of dystrophin breaks these connections, altering the plasma membrane and finally producing myofibre degeneration and necrosis.2 More precisely, dystrophin gene mutations give rise to 2 essential clinical forms of muscular dystrophy: a severe form known as Duchenne muscular dystrophy (DMD; MIM #310200), as well as a milder form, Becker muscular dystrophy (BMD; MIM#300376). Other dystrophinopathies include outliers or intermediate phenotypes, myalgia and cramp syndrome, and X-linked dilated cardiomyopathy. Less frequently, female carriers may show symptoms of varying degrees of severity.3–5
DMD is the most frequent hereditary muscular disease with an incidence estimated at 1/3500 male births.6 This disease is characterised by a complete lack of dystrophin; this situation is usually caused by mutations that alter the reading frame to produce a truncated, non-functional protein. These mutations frequently generate a premature stop codon that activates nonsense-mediated mRNA decay (NMD).7 On the other hand, BMD and other dystrophinopathies are mainly caused by mutations that do not change the reading frame of the gene and therefore generate a semi-functional protein. This phenotype-genotype correlation explains 92% of all cases and is known as the reading frame hypothesis.8
Different types of mutations have been linked to DMD. Some 65% to 70% of the cases are caused by large-scale deletions (one or more exons), whereas about 7% to 10% are caused by large duplications (one or more exons). The remaining cases of DMD/BMD are caused by point mutations (mainly nonsense mutations) and small deletions or insertions.9–12
Numerous epidemiological studies published over the years provide data on the incidence and prevalence of DMD in different populations in Sweden,13 Denmark,14 the north of England,15 Northern Ireland,16 northeastern Tuscany,17 Estonia,18 the United States,19 South Africa,20 Egypt,21 China,22 and Japan.23 Additionally, a 2014 systematic review and meta-analysis on the epidemiology of DMD estimates worldwide prevalence at 4.78/100000 male births.24
Patients with DMD are normally diagnosed between the ages of 3 and 5 years. The course of the disease is marked by progressive muscular weakness, with loss of the ability to walk at about 13 years of age. Low intellectual functioning is also present in approximately 30% of the cases.25 Other common features include heart and respiratory disease, which typically results in death between the ages of 20 and 30.26
This article summarises information on the genetic alterations present in a large group of Spanish patients diagnosed with Duchenne muscular dystrophy between 2007 and 2014. We will provide an overview of the state of this disease in Spain and revise the algorithm used in genetic diagnosis.
Patients and methodsWe reviewed a total of 284 male patients diagnosed genetically with DMD between January 2007 and December 2014. Cases were gathered from 8 Spanish referral hospitals which together provide service to most of the country: 151 cases from Hospital de la Santa Creu i Sant Pau (Barcelona), 39 from Hospital Universitario Virgen del Rocío (Seville), 38 from Hospital Universitari i Politècnic La Fe (Valencia), 26 from Hospital Universitario La Paz (Madrid), 18 from Hospital Universitario Fundación Jiménez Díaz (Madrid), and 12 from Complexo Hospitalario Universitario de Vigo (Pontevedra), Hospital Universitario de Basurto (Vizcaya), and Hospital Clínico San Carlos (Madrid). DMD was diagnosed by specialists based on strict criteria including a clinical presentation suggestive of DMD, family history of X-linked muscular dystrophy, genetic analysis of the DMD gene, and/or muscle biopsy with a dystrophin analysis performed using immunohistochemistry or Western blot.
DNA extractionGenomic DNA was extracted from peripheral blood leucocytes according to standard protocols. All patients, or patients’ guardians for minors, signed informed consent forms prior to undergoing genetic studies.
Multiplex polymerase chain reactionMultiplex polymerase chain reaction enables identification of large-scale deletions in DMD. This simultaneous amplification technique is applied to 2 series of oligonucleotides in the dystrophin gene27,28: those of the muscle promoter and those in the 17 exons in which most deletions occur (Pm, 3, 4, 6, 8, 12, 13, 17, 19, 43, 44, 45, 47, 48, 50, 51, 52, and 60). Some laboratories of reference complement the multiplex PCR study with a third series of oligonucleotides including the brain promoter and 6 additional dystrophin exons (2, 20, 33, 53, 54, and 66).
Multiplex ligation-dependent probe amplificationMultiplex ligation-dependent probe amplification (MLPA) is a technique for identifying both large-scale deletions and duplications in DMD by hybridising specific probe oligonucleotides that have been labelled and amplified by PCR. In those patients in whom multiplex PCR did not identify a deletion in the dystrophin gene, or to provide initial genetic information in some cases, researchers identified deletions and duplications using SALSA® MLPA® reagent kits P034 and P035 according to the manufacturer's instructions (MRC-Holland, Amsterdam).
SequencingResearchers detected point mutations in genomic DNA using the Sanger sequencing technique for all 79 exons and the intron regions flanking the DMD gene. For cases with available muscle tissue, molecular analysis was first performed in cDNA retrotranscribed from mRNA extracted from muscle biopsy samples; identified mutations were later confirmed in genomic DNA.
Some centres analysed point mutations with next-generation sequencing, using the MiSeq platform by Illumina, or PGM by Life Technologies.
Confirming mutationsWe revised all of the mutations recorded in this study according to the standards established by the Human Genome Variation Society (HGVS, www.hgvs.org), the Leiden Open Variant Database (LOVD, www.dmd.nl), and the Exome Variant Server (http://evs.gs.washington.edu/EVS/) to confirm current nomenclature and establish whether they had been described previously. Nucleotide positions were determined according to the reference sequence for DMD (RefSeq NM_004006.2). Pathogenicity of the new identified variations was determined using an in silico analysis involving computational prediction algorithms. We analysed alterations in splicing sites using different positional weight matrices provided by the Human Splicing Finder and Alamut programs. Alterations due to amino acid substitutions were evaluated using the Polyphen-2 and SIFT programmes.
Population data analysisPrevalence studies were carried out using data provided by Spain's National Statistics Institute with 1 January 2015 as the reference date for calculations.
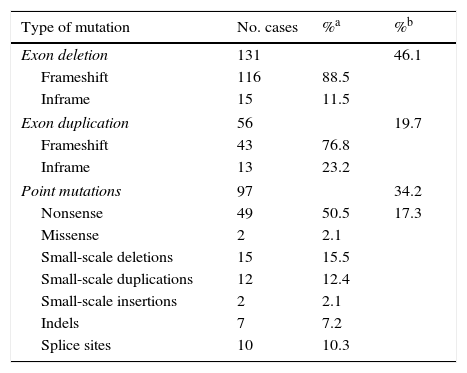
ResultsLarge-scale deletions and duplicationsBy using a combination of multiplex PCR and MLPA to perform mutation analysis of DMD in these 284 patients, we identified 187 patients as having deletions or duplications of one or more exons: 46.1% for deletions (131/284) and 19.7% for duplications (56/284) (Table 1).
Mutations identified in DMD.
| Type of mutation | No. cases | %a | %b |
|---|---|---|---|
| Exon deletion | 131 | 46.1 | |
| Frameshift | 116 | 88.5 | |
| Inframe | 15 | 11.5 | |
| Exon duplication | 56 | 19.7 | |
| Frameshift | 43 | 76.8 | |
| Inframe | 13 | 23.2 | |
| Point mutations | 97 | 34.2 | |
| Nonsense | 49 | 50.5 | 17.3 |
| Missense | 2 | 2.1 | |
| Small-scale deletions | 15 | 15.5 | |
| Small-scale duplications | 12 | 12.4 | |
| Small-scale insertions | 2 | 2.1 | |
| Indels | 7 | 7.2 | |
| Splice sites | 10 | 10.3 | |
We identified 71 different types of large-scale deletions; the most frequent was the deletion of exon 51 (10/131, 7.6%), followed by deletion of exon 45 (7/131, 5.3%), equal numbers of deletions of exons 3-7 and 45–50 (6/131, 4.6%), and deletion of exon 44 (5/131, 3.8%). Analysis of the lengths of the large-scale deletions that were identified showed that the more frequent ones affected a single exon; this occurred in 29% (38/131) of the cases caused by a large deletion. These data, and the distribution of the rest of the large-scale deletions identified in this study, confirm that mutations tended to cluster in 2 regions of the dystrophin gene: 32.7% in the region proximal to the 5′-end of the N-terminus (exons 2 to 20) and 57.4% in the medial-distal region of the central domain (exons 44 to 55). These regions are known hotspots in this gene (Fig. 1).

Regarding the effect that large-scale deletions have on transcription, 88.5% of the large deletions identified in our study (116/131) altered the reading frame. The remaining 11.5% (15/131) did not, and therefore they do not fit the reading frame hypothesis.8 In these cases, DMD was diagnosed based on clinical criteria and/or findings from muscle biopsy studies (Table 1).
We identified 27 types of large-scale duplications, the most frequent of which was duplication of exon 2 (10/56, 17.9%). As in the case of large-scale deletions, we observed that the most frequent large-scale duplications involved a single exon (15/56, 26.8%). Analysing the location of large-scale duplications also yielded a cluster in the region proximal to the 5′-end of the N-terminus (67.9%) and in the medial-distal region of the central domain (28.6%) (Fig. 1).
We analysed the frequency with which intron regions of DMD are involved in breakpoints of large-scale deletions and duplications, whether at the 5′-end or at the 3′-end. Regarding large-scale deletions, we observed that the most commonly affected regions, accounting for 64.9% of the total, are those corresponding to intron 7 (30/131), intron 44 (26/131), and intron 50 (29/131). The analysis of large-scale duplications showed that the most commonly affected regions corresponded to intron 1 (12/56), intron 2 (18/56), and intron 9 (12/56); these locations accounted for 75% of the total duplications. In contrast with large-scale deletions, in which breakpoints appear to cluster in the 2 regions displaying the most mutations, breakpoints for large-scale duplications are distributed all along the dystrophin gene (Fig. 2).
Point mutations, nonsense mutations, and others
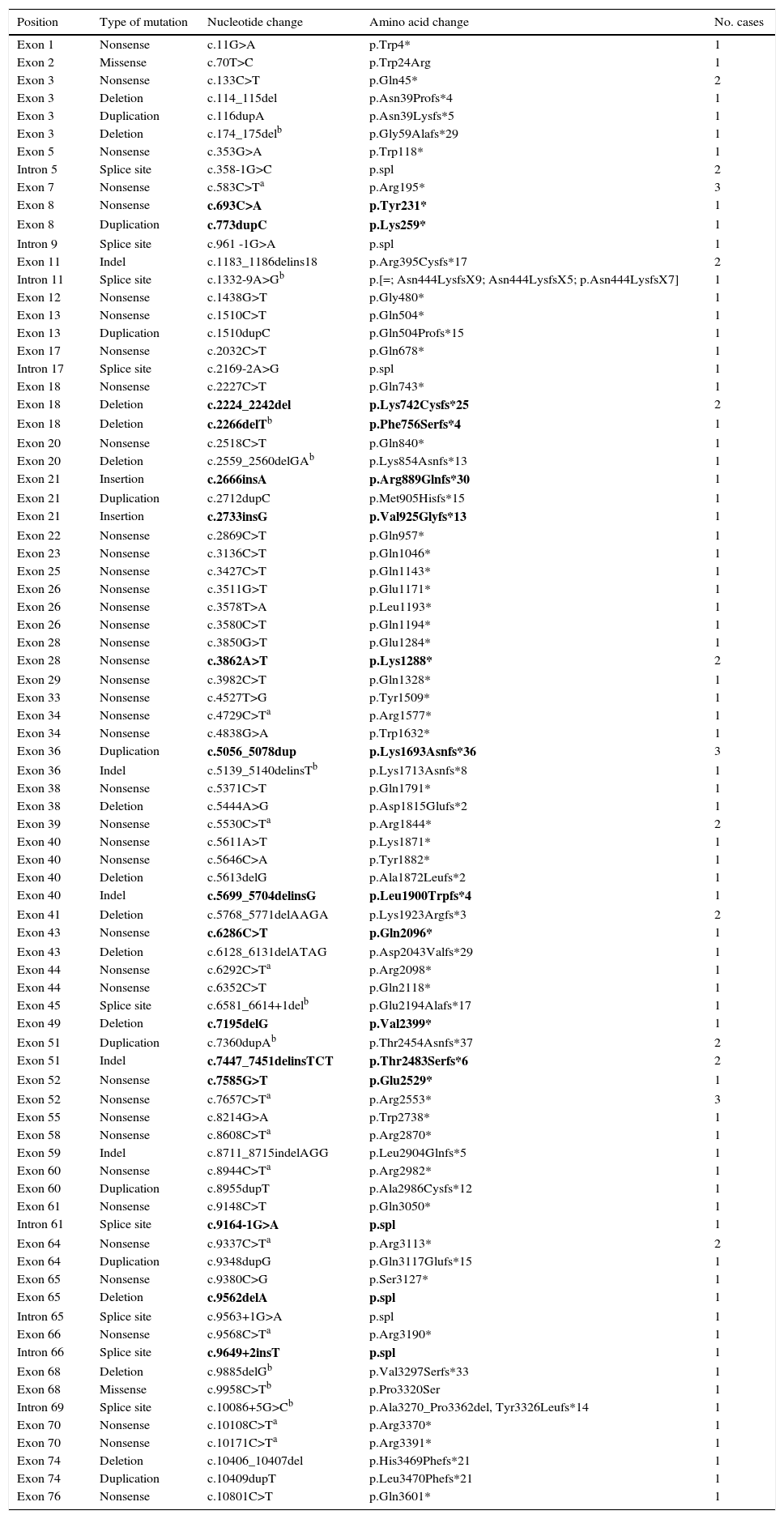
For those patients in whom multiplex PCR and MLPA did not identify the mutation in DMD, we used the Sanger sequencing technique in DNA, or in RNA where muscle biopsies were available. This analysis was performed using next-generation sequencing in more recently studied cases. We identified 97 point mutations corresponding to 34.2% of the initial 284 patients. It should be noted that more than half of these identified point mutations are nonsense mutations or nucleotide substitutions generating stop codons (49/97, 50.5%) (Table 1). In mutations of this type, cytosine was replaced by thymine in 67.3% of the cases (33/49); in 51.5% of the cases (17/33), cytosines formed part of CpG dinucleotides (Table 2). In 16 cases, the mutation was identified at the transcriptional level by analysing RNA extracted from the muscle biopsy sample.
Point mutations identified in DMD.
| Position | Type of mutation | Nucleotide change | Amino acid change | No. cases |
|---|---|---|---|---|
| Exon 1 | Nonsense | c.11G>A | p.Trp4* | 1 |
| Exon 2 | Missense | c.70T>C | p.Trp24Arg | 1 |
| Exon 3 | Nonsense | c.133C>T | p.Gln45* | 2 |
| Exon 3 | Deletion | c.114_115del | p.Asn39Profs*4 | 1 |
| Exon 3 | Duplication | c.116dupA | p.Asn39Lysfs*5 | 1 |
| Exon 3 | Deletion | c.174_175delb | p.Gly59Alafs*29 | 1 |
| Exon 5 | Nonsense | c.353G>A | p.Trp118* | 1 |
| Intron 5 | Splice site | c.358-1G>C | p.spl | 2 |
| Exon 7 | Nonsense | c.583C>Ta | p.Arg195* | 3 |
| Exon 8 | Nonsense | c.693C>A | p.Tyr231* | 1 |
| Exon 8 | Duplication | c.773dupC | p.Lys259* | 1 |
| Intron 9 | Splice site | c.961 -1G>A | p.spl | 1 |
| Exon 11 | Indel | c.1183_1186delins18 | p.Arg395Cysfs*17 | 2 |
| Intron 11 | Splice site | c.1332-9A>Gb | p.[=; Asn444LysfsX9; Asn444LysfsX5; p.Asn444LysfsX7] | 1 |
| Exon 12 | Nonsense | c.1438G>T | p.Gly480* | 1 |
| Exon 13 | Nonsense | c.1510C>T | p.Gln504* | 1 |
| Exon 13 | Duplication | c.1510dupC | p.Gln504Profs*15 | 1 |
| Exon 17 | Nonsense | c.2032C>T | p.Gln678* | 1 |
| Intron 17 | Splice site | c.2169-2A>G | p.spl | 1 |
| Exon 18 | Nonsense | c.2227C>T | p.Gln743* | 1 |
| Exon 18 | Deletion | c.2224_2242del | p.Lys742Cysfs*25 | 2 |
| Exon 18 | Deletion | c.2266delTb | p.Phe756Serfs*4 | 1 |
| Exon 20 | Nonsense | c.2518C>T | p.Gln840* | 1 |
| Exon 20 | Deletion | c.2559_2560delGAb | p.Lys854Asnfs*13 | 1 |
| Exon 21 | Insertion | c.2666insA | p.Arg889Glnfs*30 | 1 |
| Exon 21 | Duplication | c.2712dupC | p.Met905Hisfs*15 | 1 |
| Exon 21 | Insertion | c.2733insG | p.Val925Glyfs*13 | 1 |
| Exon 22 | Nonsense | c.2869C>T | p.Gln957* | 1 |
| Exon 23 | Nonsense | c.3136C>T | p.Gln1046* | 1 |
| Exon 25 | Nonsense | c.3427C>T | p.Gln1143* | 1 |
| Exon 26 | Nonsense | c.3511G>T | p.Glu1171* | 1 |
| Exon 26 | Nonsense | c.3578T>A | p.Leu1193* | 1 |
| Exon 26 | Nonsense | c.3580C>T | p.Gln1194* | 1 |
| Exon 28 | Nonsense | c.3850G>T | p.Glu1284* | 1 |
| Exon 28 | Nonsense | c.3862A>T | p.Lys1288* | 2 |
| Exon 29 | Nonsense | c.3982C>T | p.Gln1328* | 1 |
| Exon 33 | Nonsense | c.4527T>G | p.Tyr1509* | 1 |
| Exon 34 | Nonsense | c.4729C>Ta | p.Arg1577* | 1 |
| Exon 34 | Nonsense | c.4838G>A | p.Trp1632* | 1 |
| Exon 36 | Duplication | c.5056_5078dup | p.Lys1693Asnfs*36 | 3 |
| Exon 36 | Indel | c.5139_5140delinsTb | p.Lys1713Asnfs*8 | 1 |
| Exon 38 | Nonsense | c.5371C>T | p.Gln1791* | 1 |
| Exon 38 | Deletion | c.5444A>G | p.Asp1815Glufs*2 | 1 |
| Exon 39 | Nonsense | c.5530C>Ta | p.Arg1844* | 2 |
| Exon 40 | Nonsense | c.5611A>T | p.Lys1871* | 1 |
| Exon 40 | Nonsense | c.5646C>A | p.Tyr1882* | 1 |
| Exon 40 | Deletion | c.5613delG | p.Ala1872Leufs*2 | 1 |
| Exon 40 | Indel | c.5699_5704delinsG | p.Leu1900Trpfs*4 | 1 |
| Exon 41 | Deletion | c.5768_5771delAAGA | p.Lys1923Argfs*3 | 2 |
| Exon 43 | Nonsense | c.6286C>T | p.Gln2096* | 1 |
| Exon 43 | Deletion | c.6128_6131delATAG | p.Asp2043Valfs*29 | 1 |
| Exon 44 | Nonsense | c.6292C>Ta | p.Arg2098* | 1 |
| Exon 44 | Nonsense | c.6352C>T | p.Gln2118* | 1 |
| Exon 45 | Splice site | c.6581_6614+1delb | p.Glu2194Alafs*17 | 1 |
| Exon 49 | Deletion | c.7195delG | p.Val2399* | 1 |
| Exon 51 | Duplication | c.7360dupAb | p.Thr2454Asnfs*37 | 2 |
| Exon 51 | Indel | c.7447_7451delinsTCT | p.Thr2483Serfs*6 | 2 |
| Exon 52 | Nonsense | c.7585G>T | p.Glu2529* | 1 |
| Exon 52 | Nonsense | c.7657C>Ta | p.Arg2553* | 3 |
| Exon 55 | Nonsense | c.8214G>A | p.Trp2738* | 1 |
| Exon 58 | Nonsense | c.8608C>Ta | p.Arg2870* | 1 |
| Exon 59 | Indel | c.8711_8715indelAGG | p.Leu2904Glnfs*5 | 1 |
| Exon 60 | Nonsense | c.8944C>Ta | p.Arg2982* | 1 |
| Exon 60 | Duplication | c.8955dupT | p.Ala2986Cysfs*12 | 1 |
| Exon 61 | Nonsense | c.9148C>T | p.Gln3050* | 1 |
| Intron 61 | Splice site | c.9164-1G>A | p.spl | 1 |
| Exon 64 | Nonsense | c.9337C>Ta | p.Arg3113* | 2 |
| Exon 64 | Duplication | c.9348dupG | p.Gln3117Glufs*15 | 1 |
| Exon 65 | Nonsense | c.9380C>G | p.Ser3127* | 1 |
| Exon 65 | Deletion | c.9562delA | p.spl | 1 |
| Intron 65 | Splice site | c.9563+1G>A | p.spl | 1 |
| Exon 66 | Nonsense | c.9568C>Ta | p.Arg3190* | 1 |
| Intron 66 | Splice site | c.9649+2insT | p.spl | 1 |
| Exon 68 | Deletion | c.9885delGb | p.Val3297Serfs*33 | 1 |
| Exon 68 | Missense | c.9958C>Tb | p.Pro3320Ser | 1 |
| Intron 69 | Splice site | c.10086+5G>Cb | p.Ala3270_Pro3362del, Tyr3326Leufs*14 | 1 |
| Exon 70 | Nonsense | c.10108C>Ta | p.Arg3370* | 1 |
| Exon 70 | Nonsense | c.10171C>Ta | p.Arg3391* | 1 |
| Exon 74 | Deletion | c.10406_10407del | p.His3469Phefs*21 | 1 |
| Exon 74 | Duplication | c.10409dupT | p.Leu3470Phefs*21 | 1 |
| Exon 76 | Nonsense | c.10801C>T | p.Gln3601* | 1 |
Previously undescribed point mutations shown in bold.
The rest of the identified mutations account for 16.9% of the patient total; they include 15 small deletions, 12 small duplications, 10 splicing changes, 7 indels, 2 small insertions, and 2 missense substitutions. These variations range in size from a single nucleotide substitution to the deletion of 23 nucleotides. In 10 cases, the mutation was confirmed at the transcription level (Table 2).
New mutations in the DMD geneThis study identified 23 previously undescribed alterations in the DMD gene. Seven are large-scale deletions (deletion of exons 2 to 15; 3 to 45; 56 to 61; 56 to 64; 63 to 64; 64 to 70; and deletion of exon 67). Sixteen are point mutations: 4 nonsense substitutions, 4 small-scale deletions, 2 small-scale duplications, 2 indels, 2 small insertions, and 2 splicing variants (Table 2).
Geographic distribution of study casesThis case series represents approximately 26% of all of the DMD cases (284/1091) projected to have been present in Spain during the study period, based on an estimated global prevalence of DMD of 4.78 cases (95% CI, 1.94-11.81) per 100000 male population24 and population data provided by the National Statistics Institute (male population of 22820775 in January 2015). The cases observed in this study present a heterogeneous geographical distribution. The regions with the most genetically confirmed cases of DMD are those containing the hospitals of reference that participated in this study: Catalonia, Madrid, Andalusia, the Valencian Community, Basque Country, Galicia, and Cantabria.
DiscussionDMD is one of the most common neuromuscular diseases to show an X-linked inheritance pattern. It represents the most severe phenotypic variant associated with the dystrophin gene and is usually diagnosed between the ages of 2 and 5. The lack of dystrophin results in progressive degeneration of muscle fibre leading to end-stage fibrotic and adipose substitution, causing progressive muscle weakness and loss of the ability to walk by about age 13. Unlike in other dystrophinopathies, the clinical course of patients affected by DMD is relatively uniform. Differences in how individual cases progress are mostly due to associated heart disease and any personalised therapies and care the patient receives, such as physical therapy, cardiac and respiratory care, orthopaedics, and psychosocial and nutritional support.14,29,30
This article presents the cases of DMD that have been diagnosed by leading Spanish molecular genetics laboratories in order to gain a more detailed view of their genetic substrates. Mutation analysis of the 284 patients in the study confirms a high degree of genetic heterogeneity for DMD. The 179 different mutations identified in the DMD gene can be placed in 3 categories: large-scale deletions (71 different types in 131 patients); large-scale duplications (27 different types in 56 patients); and point mutations (81 different types in 97 patients).
In this series of patients with DMD, 41.6% of the cases presented large-scale deletions. This percentage is lower than that reported by earlier studies.31 This difference could reflect a selection bias, given that some of the patients were carriers of point mutations that could not be detected before 2007. These patients underwent an additional analysis at a later date, using new technologies.11 The most frequent deletions in our series affected exons 51 (7.6%) and 45 (5.3%). Deletions were clustered in 2 hotspots in this gene: 38.2% in the proximal region and 55.9% in the distal region. These data are consistent with results from other large series of patients with DMD.11,12,32,33
Exon duplications were present in 19.7% of the series, and 17.9% of them carried a duplication of exon 2. These data are also consistent with those from earlier studies.12,34 An analysis of the distribution of the large-scale duplications revealed clustering in the dystrophin gene hotspots. However, this series showed a larger cluster in the proximal region than in the distal region (67.9% and 28.6%, respectively). Again, these data are in line with results from prior studies.11,12,32,33
The distribution patterns for breakpoints for mutations differed between large-scale deletions and duplications. Breakpoints for deletions were concentrated in dystrophin hotspots, especially in the distal region; in contrast, the breakpoints applying to duplications were mainly located in the proximal region, with the rest scattered across the length of the gene. On the other hand, although the breakpoints in deletions essentially coincide with large introns, the length of the intron region should not be regarded as the sole factor to determine the location of deletion breakpoints in DMD.35 In our series, intron 7 measuring 110kb presents the highest number of breakpoints, followed by intron 50, which is much smaller at 46kb.
Contrasting with large-scale deletions and duplications, point mutations, which together account for 34.2% of the case series, are distributed evenly along the full length of DMD and there are very few coincidences; most of these mutations were identified in just one patient each. Nonsense substitutions are the most frequent type of point mutation, and they were present in 50.5% of the cases in this series. One relevant observation was the high percentage of cytosine to thymine substitutions, corresponding to the cytosines appearing in CpG dinucleotides (51.5%). Furthermore, dystrophin presents 29 CGA codons (arginine), and our study identified cytosine to thymine substitutions in 11 of them. Several earlier studies have also remarked on the tendency for thymine to replace methylated cytosine in CpG dinucleotides by means of spontaneous deamination. In this way, they have identified these CpG dinucleotides as hotspots for mutations in the dystrophin gene.11,32,33,36,37
The reading frame hypothesis established by Monaco et al.8 postulates that mutations that change the reading frame result in the lack of dystrophin which is responsible for the DMD phenotype. In this series, 254 patients are carriers of mutations that alter the reading frame (89.4%) compared to a much smaller group of patients whose mutations do not affect the reading frame (30 patients, 10.6%). Examining only large-scale deletions shows that 88.5% are compatible with the reading frame hypothesis and 11.5% are not. One explanation for these exceptions could be the presence of the deletion breakpoint within the exon region, which could in fact result in an altered reading frame.38 On the other hand, some deletions that do not change the reading frame are able to affect the dystrophin domains (actin-binding and cysteine-rich domains), which would severely impair the functionality of this protein.33 We must be mindful that applying this hypothesis to duplications is not completely reliable; it assumes that a tandem duplication is present, but this is not always the case and an mRNA study is needed to confirm that assumption.34 Missense mutations constitute another type that does not fit the frame-shift hypothesis; in theory, they are associated with less severe dystrophinopathy phenotypes. In contrast, the 2 mutations of this type that were identified in our case series were located in dystrophin domains: the mutation p.Trp24Arg is located in the ABD domain, and the p.Pro3320Ser in the CRD domain. Earlier mRNA studies have shown that splicing changes also tend to alter the reading frame.11
Based on the results obtained, we believe that the diagnostic algorithm used in hospitals participating in the study is the best able to deliver a molecular diagnosis of DMD. By using MLPA and/or multiplex PCR, it yields the genotype of 65.8% of the cases; in the secondary analysis, it uses sequencing technologies to complete the analysis of point mutations in DMD.
Recent years have seen the development of numerous clinical trials revolving around treatments for patients with DMD. They include strategies for gene substitution, correction of the DMD gene, or modification of the gene product, and they are only applied to patients carrying specific types of mutations.39,40 One of the most promising treatments being researched at present achieves readthrough of nonsense mutations. In this process, treatment with PTC124 suppresses the effect of substitutions that generate premature stop codons.41,42 Carriers of nonsense mutations make up 17.2% of the DMD cases in our series (110/284), and they may benefit from this type of treatment. Another noteworthy line of treatment for patients with DMD employs exon skipping, which is intended to re-establish the reading frame by suppressing one or more exons. The mechanism for skipping exon 51 is the most developed to date, followed by those for exons 44, 45, 52, 53, and 55.43,44 The genetic analysis revealed that 21.5% of these patients (61/284) presented deletions, but that the reading frame for dystrophin could be re-established by omitting one of the above exons. As a result, 38.7% of the patients in our case series (110/284) would theoretically be eligible to participate in one of these recent clinical trials, depending on further inclusion/exclusion criteria. With this reflection, we wish to highlight the transcendence of being able to identify the type of mutation in patients with DMD, since it can help them be selected for different clinical trials. On the other hand, genetic diagnosis is equally important as a means of preventing further cases in an affected family.
One of the objectives of this article was to analyse the geographic distribution of patients with DMD in Spain between 2007 and 2014. We were only able to gather 26% of the cases that we might have expected to find given the Spanish population. Apart from potential cases that have not been identified, this figure stresses that DMD is a genetically underdiagnosed condition. This situation may arise in part from the difficulty of gaining access to hospitals of reference; it may also reflect insufficient communication between clinicians and geneticists. Better coordination between these specialties may help promote and improve genetic diagnosis of dystrophinopathies and other neuromuscular diseases, delivering more in-depth understanding of the clinicopathological and genetic correlation. Cooperative efforts will also favour use of potential treatments in these patients who, in the face of severe illness, have had so few treatment options until only recently.
Conflicts of interestThe authors have no conflicts of interest to declare.
The authors would like to thank Dr Susana Teijeira (Complexo Hospitalario Universitario de Vigo, Institute of Biomedical Research of Ourense-Pontevedra-Vigo [IBI]) and Dr Paloma Martínez Montero (Institute of Medical and Molecular Genetics [INGEMM], Hospital Universitario La Paz, Madrid) for their contributions to this study, as well as all participating hospitals.
Please cite this article as: Vieitez I, Gallano P, González-Quereda L, Borrego S, Marcos I, Millán JM, et al. Espectro mutacional de la distrofia muscular de Duchenne en España: estudio de 284 casos. Neurología. 2017;32:377–385.
