



To describe the history of the discovery of SCA36 and review knowledge of this entity, which is currently the most prevalent hereditary ataxia in Galicia (Spain) owing to a founder effect.
DevelopmentSCA36 is an autosomal dominant hereditary ataxia with late onset and slow progression. It presents with cerebellar ataxia, sensorineural hearing loss, and discrete motor neuron impairment (tongue atrophy with denervation, discrete pyramidal signs). SCA36 was first described in Japan (Asida River ataxia) and in Galicia (Costa da Morte ataxia). The condition is caused by a genetic mutation (intronic hexanucleotide repeat expansion) in the NOP56 gene on the short arm of chromosome 20 (20p13). Magnetic resonance image study initially shows cerebellar vermian atrophy that subsequently extends to the rest of the cerebellum and finally to the pontomedullary region of the brainstem without producing white matter lesions. Peripheral nerve conduction velocities are normal, and sensorimotor evoked potential studies show delayed conduction of stimuli to lower limbs. In patients with hearing loss, audiometric studies show a drop of >40dB in frequencies exceeding 2500Hz. Auditory evoked potential studies may also show lack of waves I and II.
ConclusionsCosta da Morte ataxia or SCA36 is the most prevalent SCA in the Spanish region of Galicia. Given the region's history of high rates of emigration, new cases may be diagnosed in numerous countries, especially in Latin America. Genetic studies are now available to patients and asymptomatic carriers. Since many people are at risk for this disease, we will continue our investigations aimed at elucidating the underlying pathogenic molecular mechanisms and discovering effective treatment.
Describir la historia del descubrimiento de la SCA36 y revisar los conocimientos actuales sobre esta entidad que, por un efecto fundador, ha pasado a ser la SCA más prevalente en Galicia (España).
DesarrolloLa SCA36 es una enfermedad heredodegenerativa autosómica dominante, de inicio tardío y lenta progresión, que cursa con ataxia cerebelosa, hipoacusia neurosensorial y discreta afectación de neuronas motoras (atrofia y fasciculaciones linguales y signos piramidales leves). Ha sido descrita inicialmente en Japón (ataxia del río Asida) y en Galicia (ataxia da Costa da Morte). Se produce por una mutación (expansión intrónica de hexanucleótido) en el gen NOP56, localizado en 20p13. En los estudios de resonancia magnética se observa inicialmente atrofia vermiana superior, que se extenderá al resto del cerebelo y finalmente a la porción bulboprotuberancial del tronco cerebral, sin lesiones de sustancia blanca. Las velocidades de conducción nerviosa periférica son normales y en los estudios de potenciales evocados somatosensoriales se detecta retraso de la conducción al estimular en miembros inferiores. En los pacientes con hipoacusia, suele encontrarse en la audiometría una caída>40dB a partir de 2.400Hz; también se observa ausencia de ondas i y ii en los estudios de potenciales evocados auditivos.
ConclusionesLa ataxia da Costa da Morte-SCA36 es la SCA más prevalente en Galicia (España). Dada la alta tasa de emigración de nuestra comunidad autónoma, se espera que se diagnostiquen nuevos casos en diversas latitudes, sobre todo en América Latina. Ahora está disponible el diagnóstico genético para pacientes con clínica y portadores asintomáticos. Dado el alto número de pacientes en riesgo de sufrir la enfermedad, continuamos con las investigaciones para aclarar los mecanismos moleculares patogénicos y poder encontrar una terapéutica.
In 1864, Duchenne de Boulogne used the term ‘locomotor ataxia’ to describe gait abnormalities caused by tabes dorsalis.1 In 1893, Pierre Marie drew a distinction between autosomal dominant hereditary ataxias and autosomal recessive ataxia, which had already been described by Nikolaus Friedreich in 1863.2 Throughout the twentieth century, Holmes, Dejerine, Greenfield, and Thomas, among others, classified degenerative ataxias based on clinical and pathological criteria into cerebellar ataxia, olivopontocerebellar atrophy, and spinocerebellar ataxia; each category was further subdivided into sporadic and hereditary.3–5 In 1980, Anita Harding6 classified autosomal dominant cerebellar ataxias (ADCA) into 3 groups: ADCA I (cerebellar syndrome sometimes accompanied by ophthalmoplegia, dementia, movement disorders, amyotrophy, or optic neuritis), ADCA II (cerebellar syndrome with retinal degeneration), and ADCA III (pure cerebellar syndrome). Harding's classification remains very useful for the initial assessment of patients with ataxia and an autosomal dominant inheritance pattern.
The first loci linked to specific ADCA types began to be identified after 19907,8 and that acronym was replaced by SCA (spinocerebellar ataxia). This term is used at present to describe a heterogeneous and expanding group of degenerative disorders with manifestations including ataxia and which follow an autosomal dominant inheritance pattern. Although 40 types of SCA have been described, only 34 loci have been identified to date: SCA9 is uncertain, SCA15 and SCA16 are the same disease, SCA24 has an autosomal recessive inheritance pattern, and no published evidence is available for SCA33.9–13
To a certain extent, clinical heterogeneity of SCA reflects the wide range of genetic alterations causing them. The types of mutations known to date are: 1) point mutations (SCA13, 23, 27, 35, 38, and 40), 2) frameshift mutations (SCA11), 3) deletions (SCA15 and SCA27), 4) duplications (SCA20), 5) CAG triplet repeat expansions in coding regions (SCA1, 2, 3, 7, 17, and DRPLA), and 6) tri-, penta-, and hexa-nucleotide repeat expansions in non-coding regions (SCA8, 10, 12, 31, and 36). Only one linkage analysis in a specific chromosomal region has been conducted for some types of SCA (SCA4, 10, 18, 19, 21, 22, 25, 26, 29, 32, 34, and 37), and the gene and type of mutation in each case is not known.9–13
This article reviews current knowledge of spinocerebellar ataxia 36 (SCA36), the latest type of SCA to be linked to a genetic mutation. We propose the alternative term ‘Costa da Morte ataxia’ after finding a high prevalence of SCA36 in this section of the Atlantic coast in Galicia, Spain (Fig. 1). We studied 2 large families with high numbers of affected patients (over 40) and carriers (over 60). According to these findings, SCA36 is the most prevalent type of SCA in Galicia. As this region has a history of intense emigration, SCA36 may also have spread to numerous other areas.
DevelopmentThe discovery of SCA
Since the early 1990s, Complexo Hospitalario Universitario de Santiago has attended numerous patients from Muxia and Ponteceso, 2 villages along the Costa da Morte (A Coruña, Spain) (Fig. 1). All displayed late-onset, slowly-progressing ataxia, and they reported similar cases among their relatives. Based on the data provided by the patients themselves, the syndrome followed an autosomal dominant inheritance pattern. As genetic studies of the first types of SCA became available, we observed that our patients did not carry the mutations or genetic linkages described in the literature. By 2005 we were well aware that all cases pertained to a relatively homogeneous disease, with a high concentration of patients in the area mentioned above. Neurologists and geneticists came together to conduct a thorough field study to visit patients and at-risk relatives. After subjects signed informed consent forms, we drew samples for genetic testing and created a detailed pedigree. Arias et al.14 described the main features of what we had identified as a new type of SCA at the Annual Meetings of the Galician Society of Neurology (A Coruña, 2008) and the Spanish Society of Neurology (Barcelona, 2008). In 2011, Kobayashi et al.15 were the first to report the mutation linked to SCA36, and our research group confirmed it shortly afterward.16 To date, several other cases have been reported in Spain, Italy, and Poland.
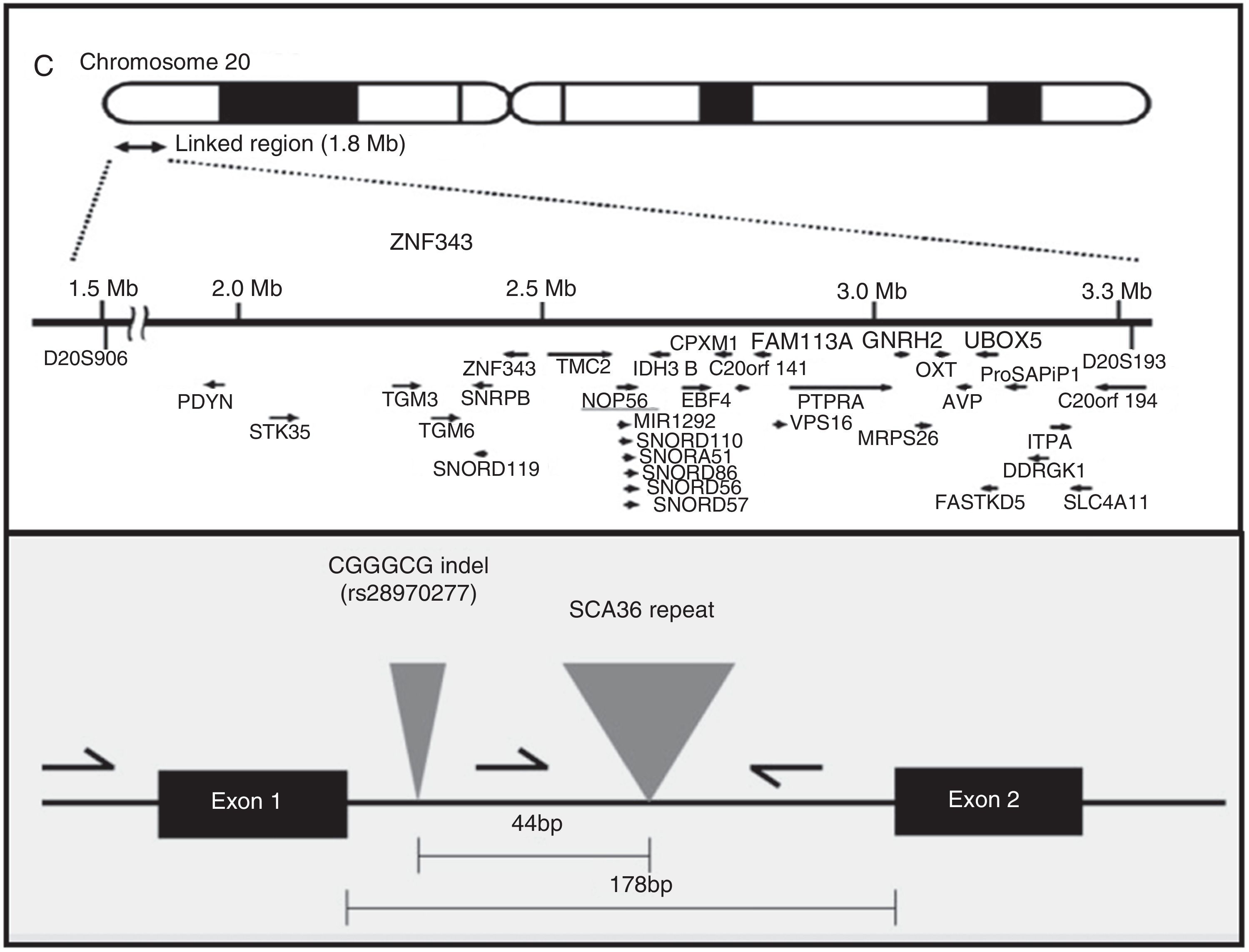
AetiopathogenesisThe NOP56 gene, located on chromosome 20p13, codes for 56-kD protein, which interacts with NOP1 and NOP58 to form the 60S ribosomal subunit. SCA36 is caused by a mutation in the NOP56 gene consisting of a heterozygous expansion of a GGCCTG hexanucleotide repeat in intron 1 (Fig. 2). Studies with lymphoblastoid cells have shown that expansions in the first intron of NOP56 produce intranuclear focal accumulation of RNA which may interfere with multiple transcription factors.15 The same occurs in other SCA types caused by expansions, as well as in Huntington disease and myotonic dystrophy. There is a growing interest in the role of RNA in neurodegenerative diseases.17
Epidemiology
Prevalence of SCA varies considerably from country to country. Generally speaking, SCA3 is the world's most frequent type of SCA; however, SCA10 is the most frequent type in Mexico, SCA7 in Scandinavian countries, and SCA2 and SCA3 in Spain.18–21 SCA36 is currently the most prevalent type of SCA in Galicia, accounting for 21.3% of all forms of autosomal dominant ataxia in adults according to the database kept by the Galician Foundation of Genomic Medicine.16 A familial connection between Galician and Japanese cases of SCA36, in addition to other cases in Spain and Europe, has yet to be confirmed. In any case, the substantial numbers of Galician emigrants who reached other parts of Spain, as well as Europe and Latin America, indicate that we may soon find new cases of SCA36 in those locations.
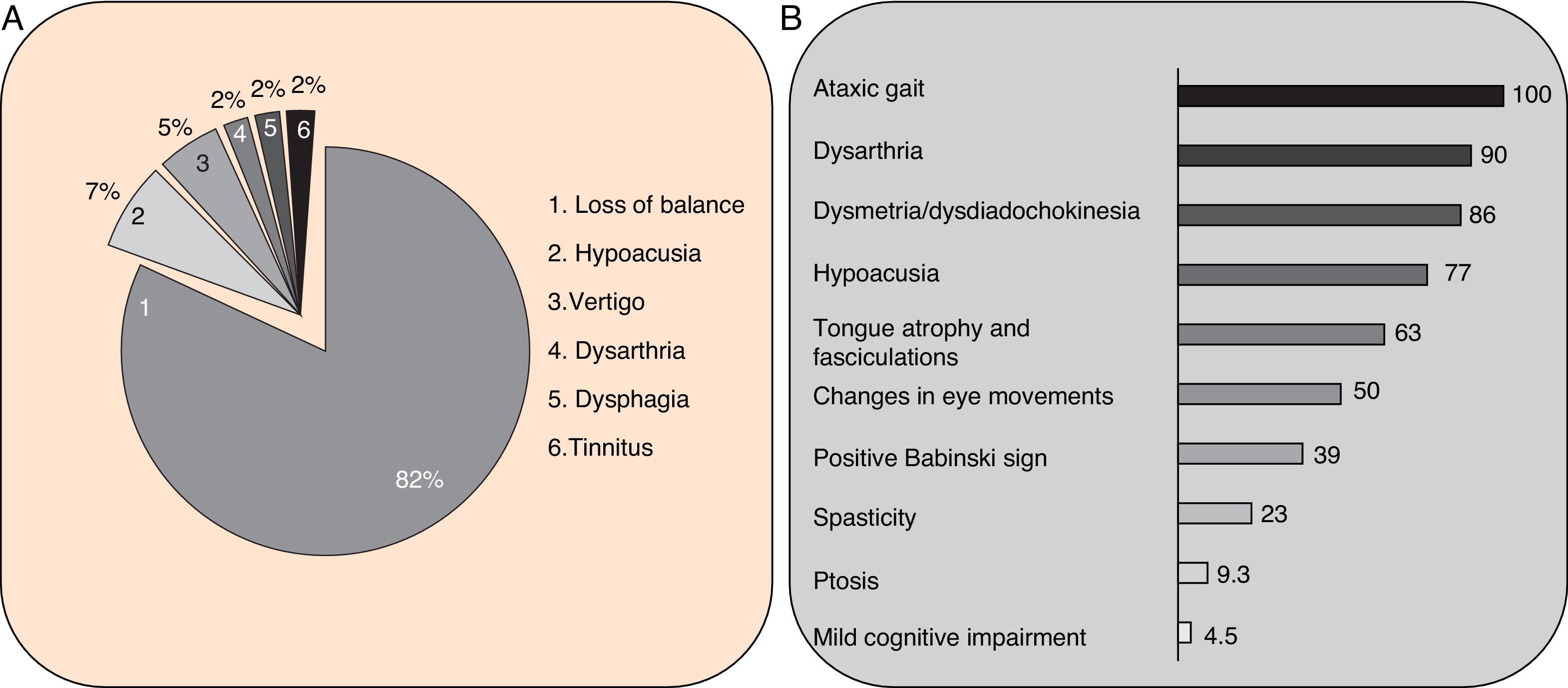
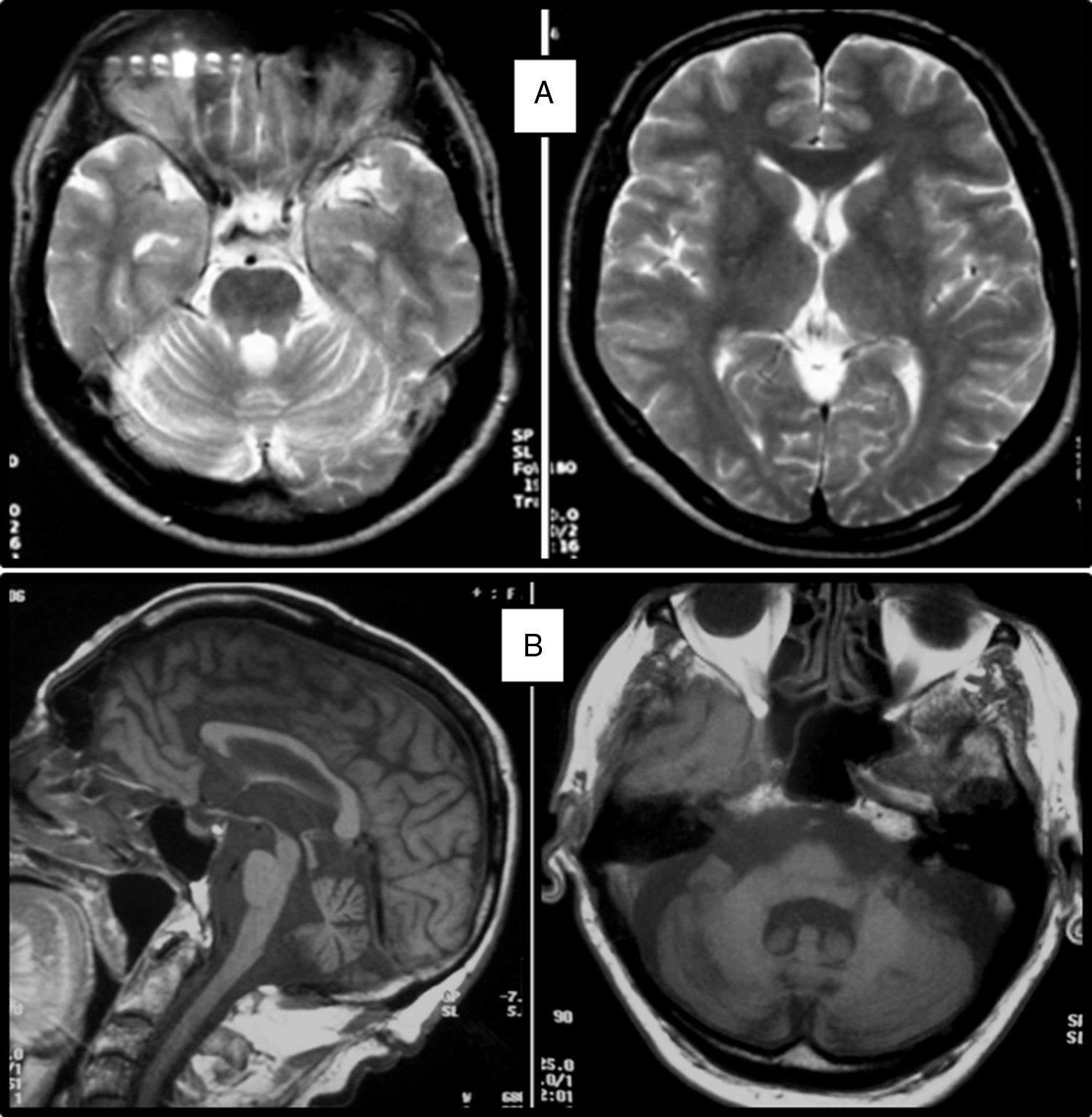
Clinical symptomsSCA36 typically starts to manifest with imbalance and instability between the ages of 40 and 60. Symptoms progress slowly; appendicular ataxia, dysmetria, dysdiadochokinesia, and cerebellar or mixed dysarthria (cerebellar and bulbar) appear at later stages.14–16,22,23 Patients rarely need a wheelchair before 15 years of disease progression. Nearly 80% of the patients display sensory hypoacusia; this symptom was not described in the first case reports of Japanese patients but has been reported in subsequent articles.15,24 Between 60% and 70% of the patients displayed tongue atrophy and fasciculations, which may contribute to dysarthria and, in some cases, to mild to moderate dysphagia (none of the published cases required a nasogastric tube or percutaneous endoscopic gastrostomy for feeding).15,16,22–26 Our study group has observed no signs or symptoms of lower motor neuron syndrome aside from those affecting the tongue; however, some cases have been reported in Japan. Over half of the cases display pyramidal symptoms, hyperreflexia, and positive Babinski sign with little to no spasticity. Other clinical signs include nystagmus, slow hypometric saccades, ptosis, and mild to moderate cognitive impairment with a pattern of frontal dysexecutive syndrome.25Fig. 3 shows the initial symptoms and manifestations associated with the disease in our series of 44 patients. None of our patients displayed dysautonomia or extrapyramidal symptoms, although Miyashiro et al.27 recently reported a case with associated parkinsonism and dystonia. The most relevant findings from complementary tests in patients with SCA3615,16 are the following: A) MRI scans reveal cerebellar atrophy as early as symptom onset; it usually starts in the superior cerebellar vermis and extends to the rest of the cerebellum down to the brainstem, displaying a pattern of olivopontocerebellar atrophy (Fig. 4) and no detectable white matter lesions. Moderate frontal atrophy may be seen in some cases. B) Neurophysiological studies reveal normal motor and sensory nerve conduction velocities; denervation in our Galician patients is limited to the tongue. Somatosensory evoked potential studies reveal conduction delays with lower limb stimulation. Auditory evoked potential studies displayed very small to no amplitude of waves I and II. Audiometry tests typically reveal a drop of 40dB or more at frequencies higher than 2500Hz. C) Although histopathology studies are rarely conducted, they reveal Purkinje cell loss, especially in the dentate nucleus, and loss of motor neurons in the hypoglossal nucleus. We did not observe the eosinophilic cytoplasmic inclusions resembling Bunina bodies and seen in motor neurons of the anterior horn of the spinal cord in patients with ALS.
 Initial symptoms in our sample of patients with SCA36. B) Clinical manifestations of SCA36 in our patients.")
 T2-weighted MRI scan of a 52-year-old patient with SCA36 showing no white matter lesions or cortical atrophy; diffuse cerebellar atrophy may be seen. B) T1-weighted MRI scan of an 86-year-old patient with SCA36 showing olivopontocerebellar atrophy.")
No clear correlation was found between early onset and severity of symptoms on the one hand, and size of the expansion on the other.14
Differential diagnosisAt onset, SCA36 resembles ADCA III (pure cerebellar syndrome); the subsequent hypoacusia and tongue fasciculations suggest a mild type of ADCA I. Differential diagnosis must therefore include SCA5, 11, 16, 26, 30, and 31. As a general rule, forms of ADCA I (SCA1, 2, and 3) all display early onset.9–13 Hypoacusia is also present in SCA31 and occasionally in other types of SCA.28 Motor neuron alterations are also found in SCA229 and dentatorubral-pallidoluysian atrophy (DRPLA). Fragile X-associated tremor/ataxia syndrome (FXTAS) has some clinical similarities to SCA36, although MRI scans in patients with FXTAS usually display typical hyperintense lesions in the middle cerebellar peduncles.30
Friedreich ataxia and such other autosomal recessive ataxias as ataxia with oculomotor apraxia types 1 and 2, ataxia-telangiectasia, and cerebrotendinous xanthomatosis may exceptionally show late onset with a mild phenotype resembling that of SCA36, although they show clinical and neurophysiological features of peripheral neuropathy31; these entities should therefore be included in the differential diagnosis in certain cases. Mitochondrial cytopathies, both those arising from mitochondrial DNA mutations (maternal inheritance) and those resulting from somatic mutations,32 are associated with a wide range of symptoms including ataxia and hypoacusia and may therefore be mistaken for SCA36. Cerebellar types of multiple system atrophy and progressive supranuclear palsy may also resemble the initial stages of SCA36, although progression and MRI findings are clearly different.
A variety of acquired late-onset ataxias may also be included in the differential diagnosis of isolated cases of SCA36: toxic (alcohol or drug use), metabolic (vitamin E deficiency and Wernicke encephalopathy), autoimmune (Miller Fisher syndrome, Bickerstaff brainstem encephalitis, coeliac disease, presence of anti-GAD or other types of antibodies), and paraneoplastic.33
Diagnosis and genetic counsellingPresence or absence of a pathogenic expansion of a GGCCTG hexanucleotide repeat in NOP56 will either confirm or rule out SCA36 in at-risk patients and in those believed to have sporadic ataxia. PCR and the appropriate primers are used to detect a heterozygous expansion. The exact size of the allele containing a large expansion can be determined with Southern blot, although this is not usually necessary. The number of CGCCTG repeats in normal alleles ranges from 3 to 14 in white individuals16,34 and from 3 to 8 in Japanese individuals.15 Expanded alleles contain over 650 repeats. Diagnosis in asymptomatic individuals, preimplantation genetic diagnosis, and prenatal diagnosis can only be performed after detecting the mutation in the family. No allelic disorders associated with a different phenotype due to NOP56 mutations have been discovered to date.
TreatmentThere is no specific treatment for SCA36. In Galicia, there are over 100 patients and carriers of the mutation (those who have voluntarily undergone genetic testing). These individuals should exercise regularly, avoid gaining weight, and not consume alcohol or other substances or medications that may be toxic to the cerebellum (phenytoin, carbamazepine, metronidazole, amiodarone, lithium) or auditory system (salicylates). They should also use earplugs to avoid acoustic trauma in case of excessive environmental noise.
Future lines of researchTo understand the molecular mechanisms of SCA36 and design treatment strategies, our research group is conducting studies along various lines of research, including epidemiology and phylogenetics of the mutation, expansion dynamics, mitotic and meiotic instability, influence of phenotypic characteristics, and effects on the transcription of other genes. Results from these investigations may usher in more effective treatment approaches, at least for asymptomatic carriers and mildly affected patients.
ConclusionsCosta da Morte ataxia is now included within SCA36 since we have detected the mutation causing the disease (expansion of intronic GGCCTG hexanucleotide repeat in NOP56). The phenotype of the disease is characterised by late-onset, slowly-progressing cerebellar syndrome; neurosensory hypoacusia; tongue fasciculations and atrophy; and mild pyramidal symptoms. This is the most frequent type of SCA in Galicia. At present, the mutation can be detected in carriers, making genetic counselling possible for individuals planning to conceive. The possibility of a familial association between Costa da Morte ataxia and Asida River ataxia (described in Japan and caused by a similar mutation) has yet to be confirmed.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Arias M, García-Murias M, Sobrido MJ. La ataxia espinocerebelosa 36 (SCA36): «Ataxia da Costa da Morte». Neurología. 2017;32:386–393.
